Help
Step 1 Choose File

After press ENCODING link you will be directed to this page.There is two options for encoding process.You can give your protein sequences as inputs or you can upload your .fasta file.
Step 2 Choose File
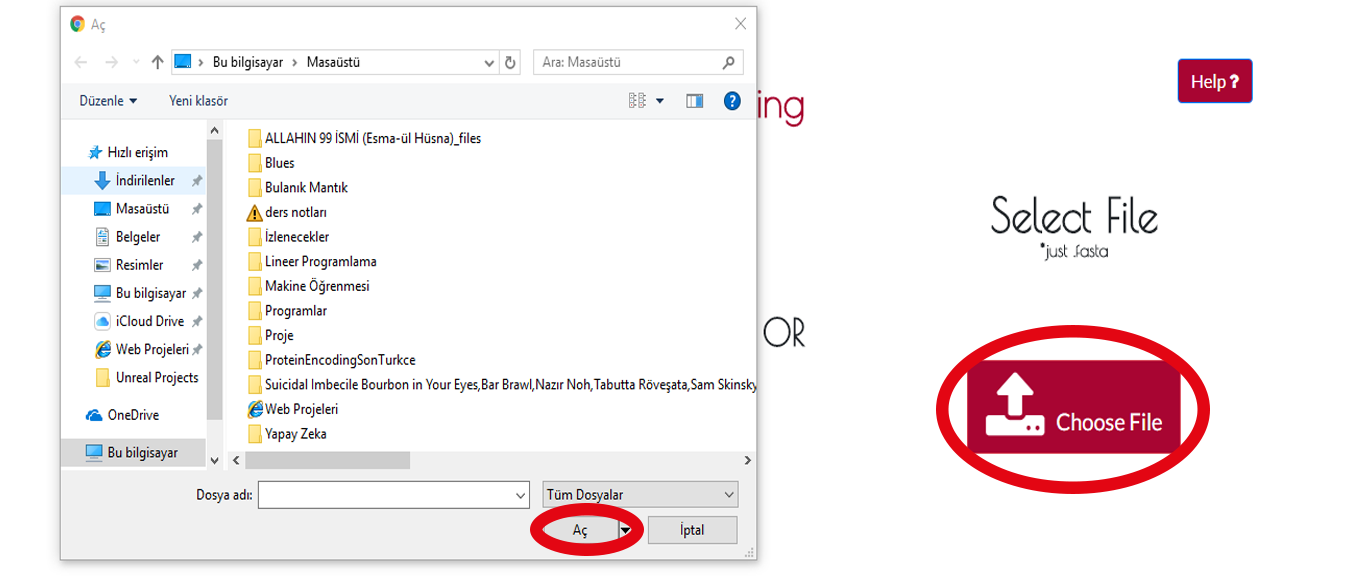
If you have a .fasta file you can upload it with pressing CHOOSE FILE button.After that a window will be opened so you can find your .fasta file.Then you will press OPEN button and that is all.
Step 1 Input Sequence

If you want to type the sequence manually you have to write it down into the textarea.If you want to type more than one sequence you need to press enter between sequences.While you are writing the sequence encoding methods and CALCULATE button will appear immediately.
Step 2 Input Sequence
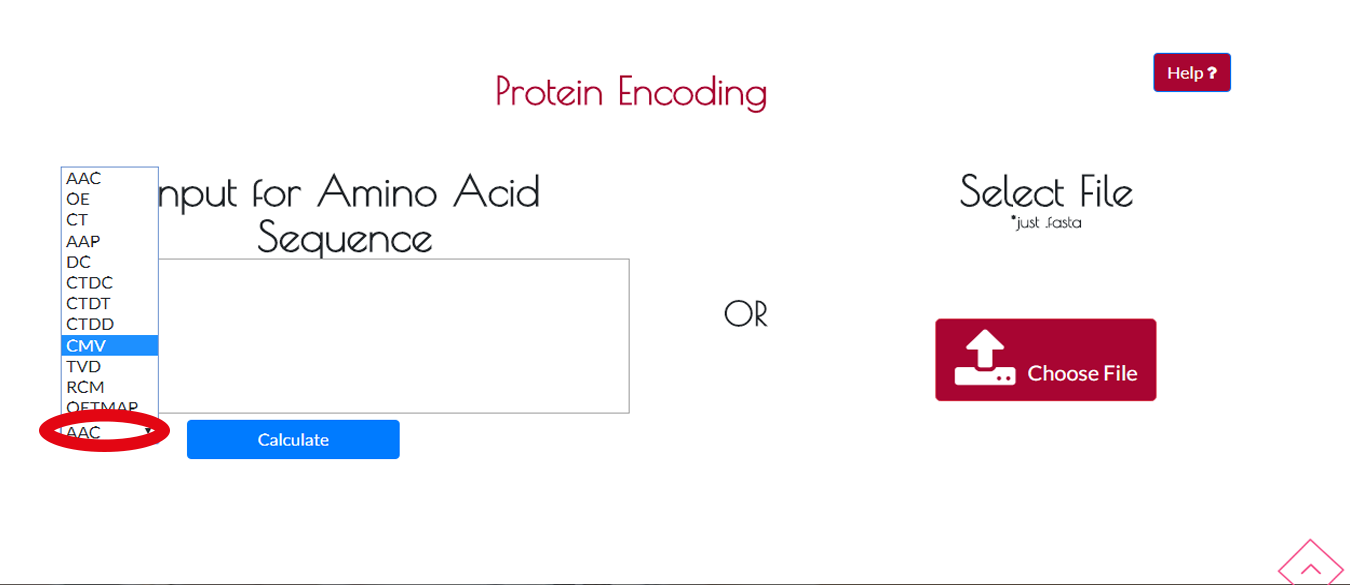
You can select the encoding method from the drop-down menu and press CALCULATE button to start the encoding process.
Step 3 Input Sequence
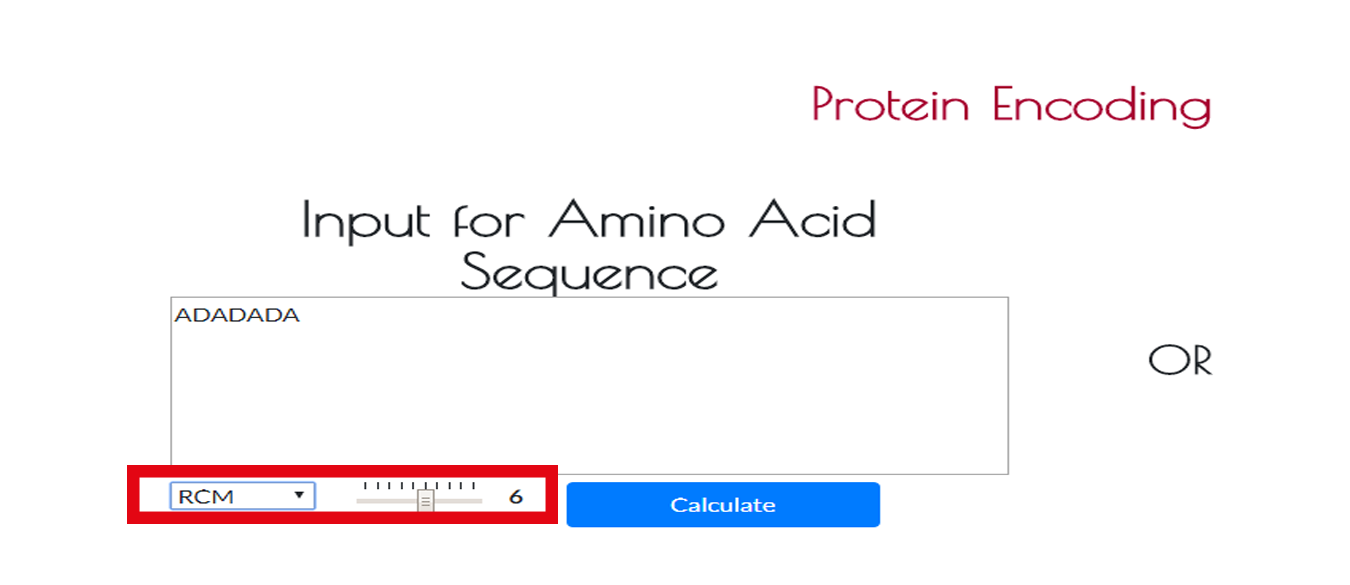
If you select RCM method, you will need to adjust rank of the RCM method.
Step 1 Select Sequence from Table
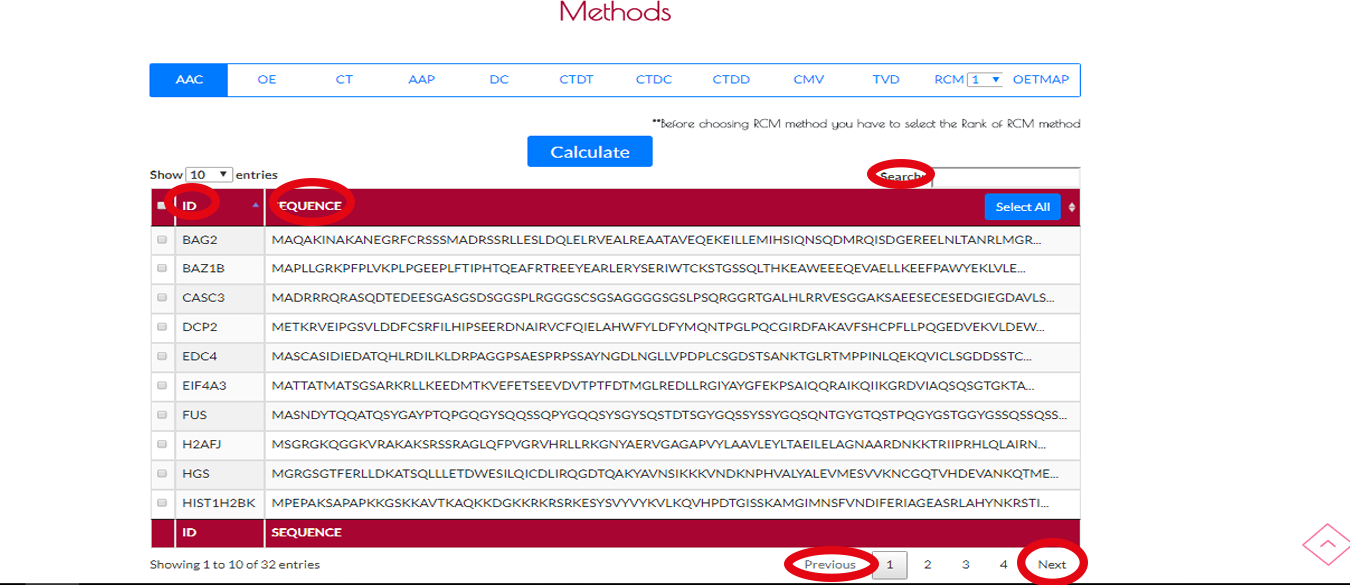
The sequence table can not be seen before choosing a file.After uploading process a table with full of sequences and IDs will appear.We have a search option on the right corner of the table.You can find your ID or sequence with that.On the bottom side of the table there are PREVIOUS and NEXT buttons.You can use these buttons to navigate on the table and see all IDs and sequences of your .fasta file.
Step 2 Select Sequence from Table
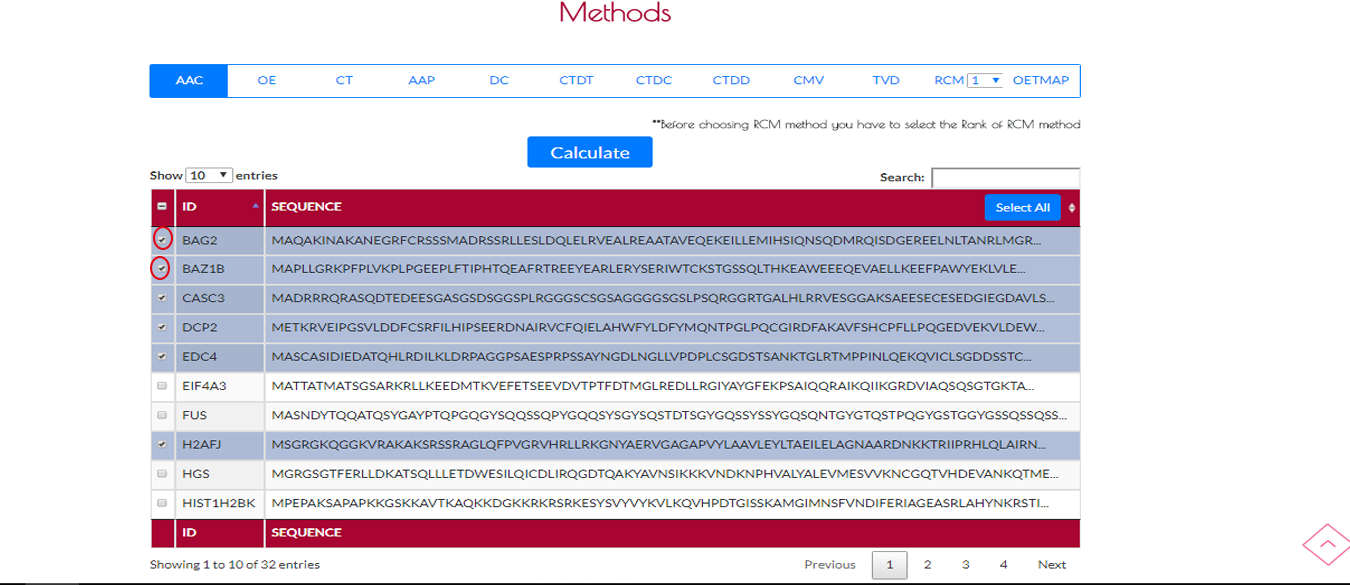
On the left side of the table there are checkboxes of sequences.You can manually choose your sequence which you want to encode.
Step 3 Select Sequence from Table
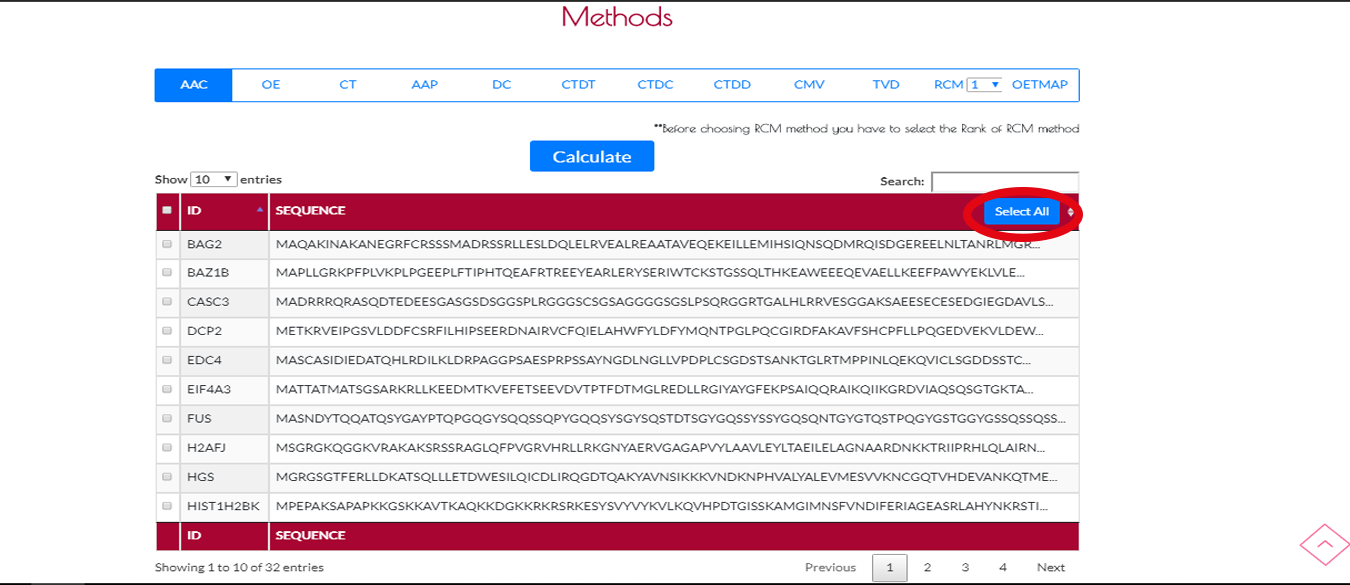
If you want to encode all the sequences of the .fasta file you have to press SELECT ALL button.If you want to select the active page then you have to check the box on the top left side of the table.
Step 4 Select Sequence from Table
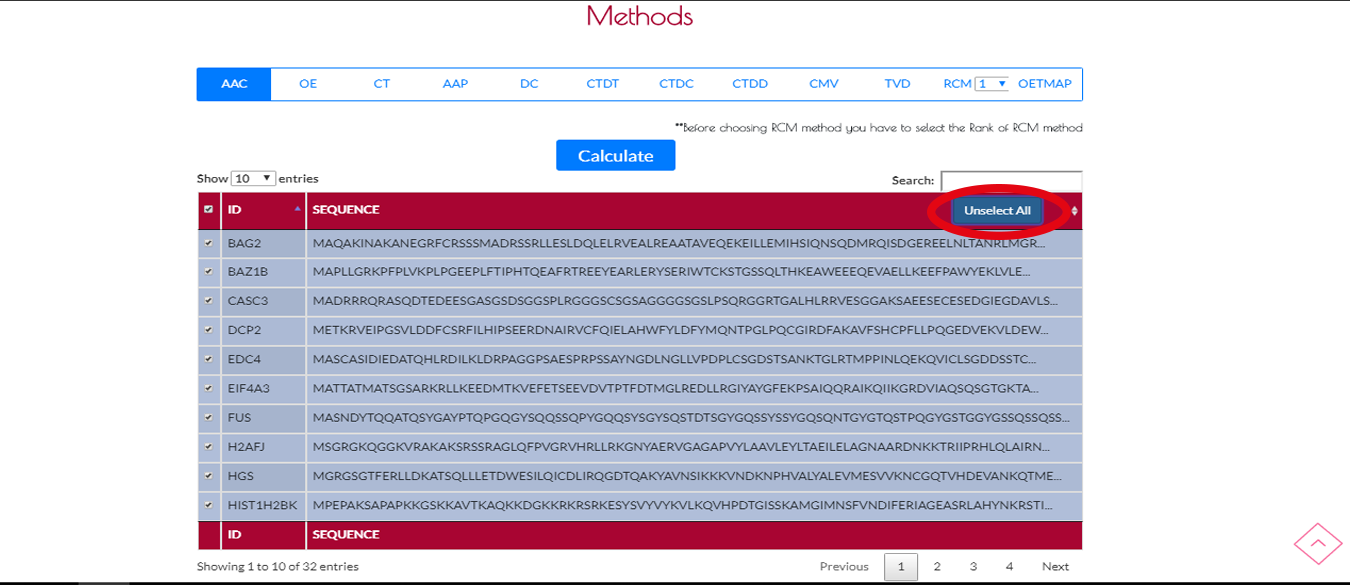
After you click SelectAll button it will change to UnselectAll.
Step 1 Calculate
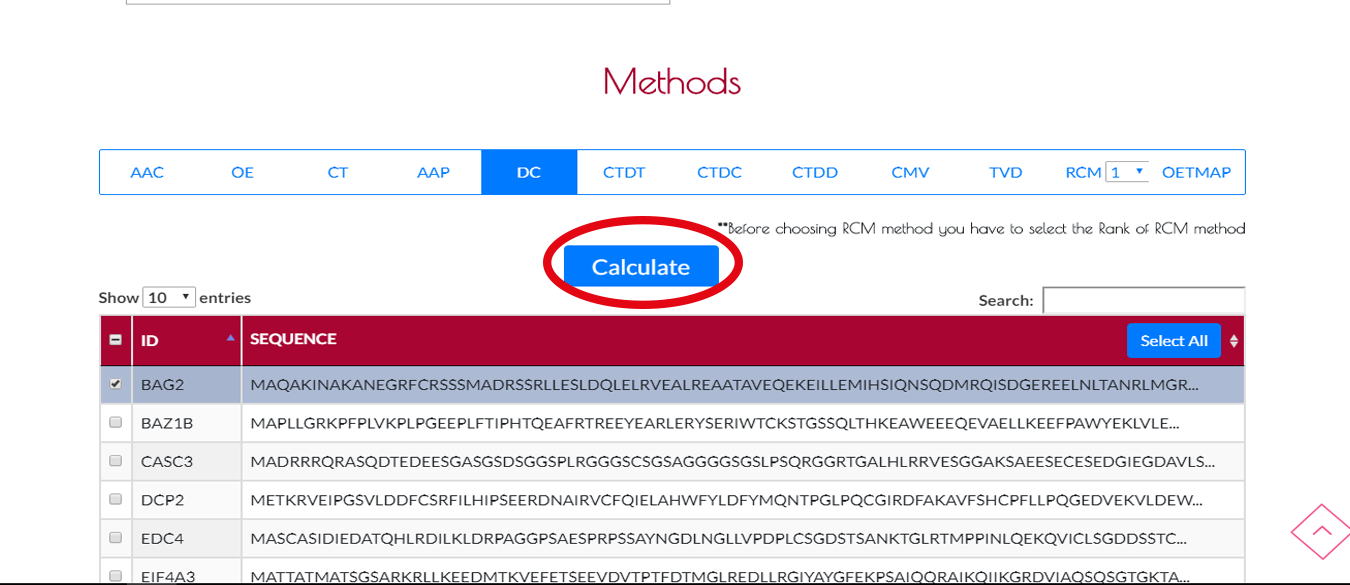
After selecting the sequences , you can choose your encoding method and press CALCULATE button to start the encoding process.
Step 2 Calculate
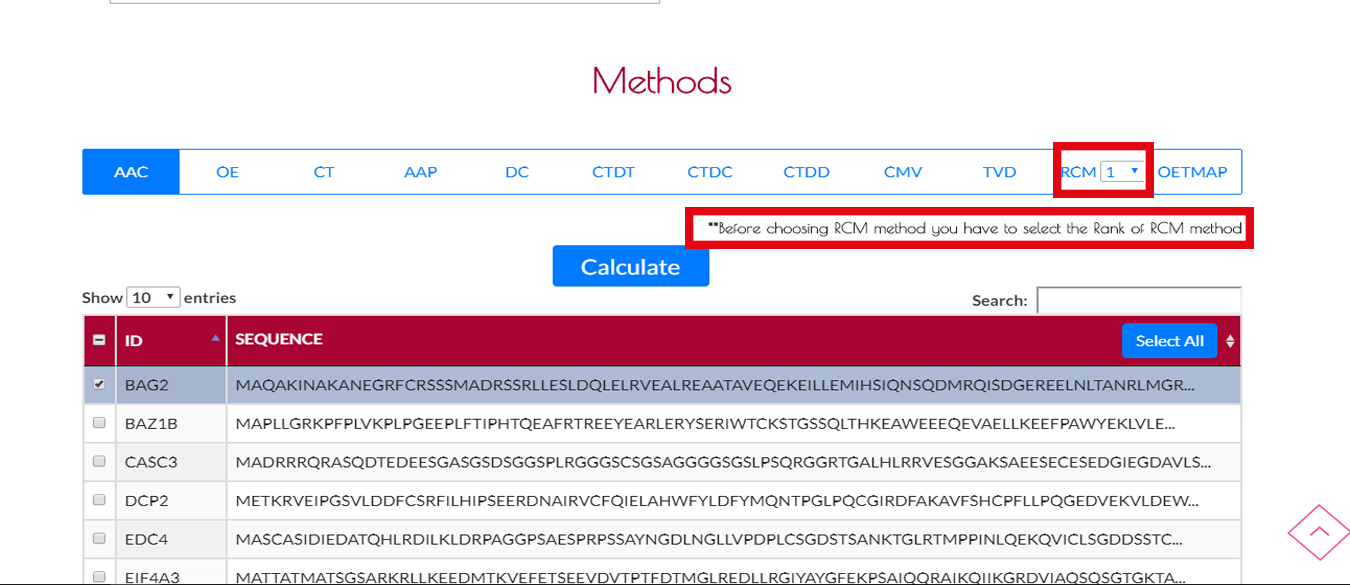
If you select RCM method, you will need to adjust rank of the RCM method.
Step 3 Calculate
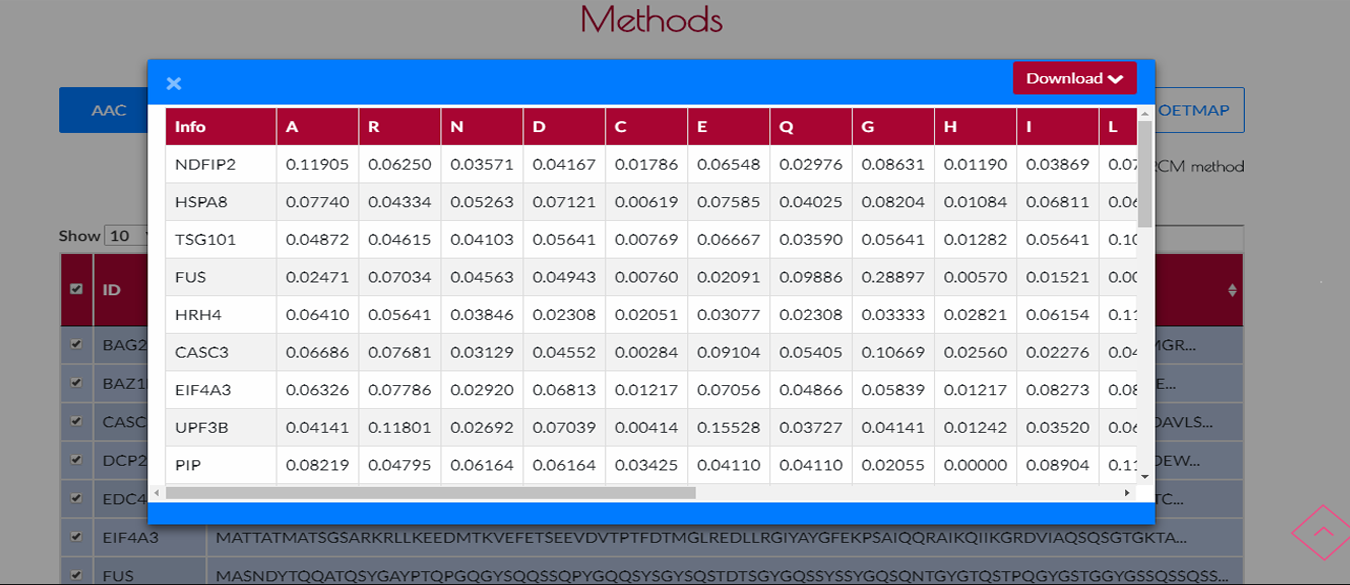
You will see the result table of your encoding method after the encoding process.
Step 4 Calculate
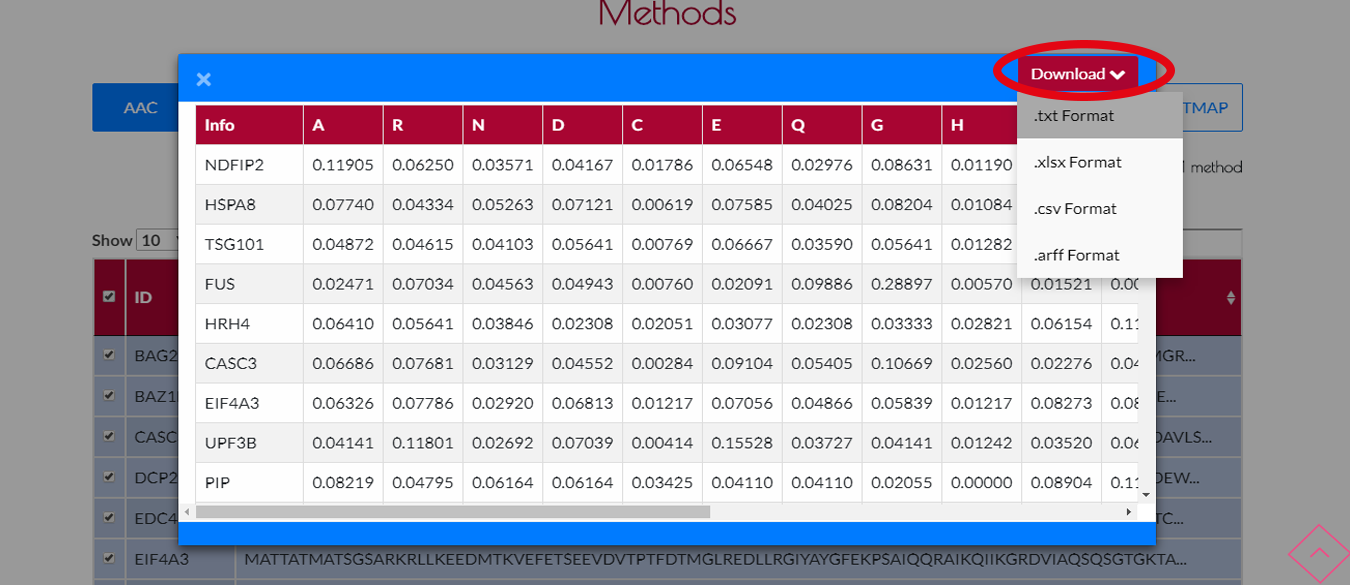
Finally if you want to download the results, you can press DOWNLOAD button.A drop-down menu will appear then you can choose your output format from this menu.(.txt,.xls,.csv,.arff) are supported formats.
Step 1 Interaction File Upload

First you have to upload your .xls or .xlsx file which can be two types of format. First format is the pathogen-host interaction data file which you dowload from phisto.org and the second format is the file which has two columns;pathogen uniprotID, host uniprotID respectively.
Step 2 Interaction File Upload
A navigation window will appear then you will find your file's path and press OPEN to finish uploading process.
Step 3 Interaction File Upload(Table Form)
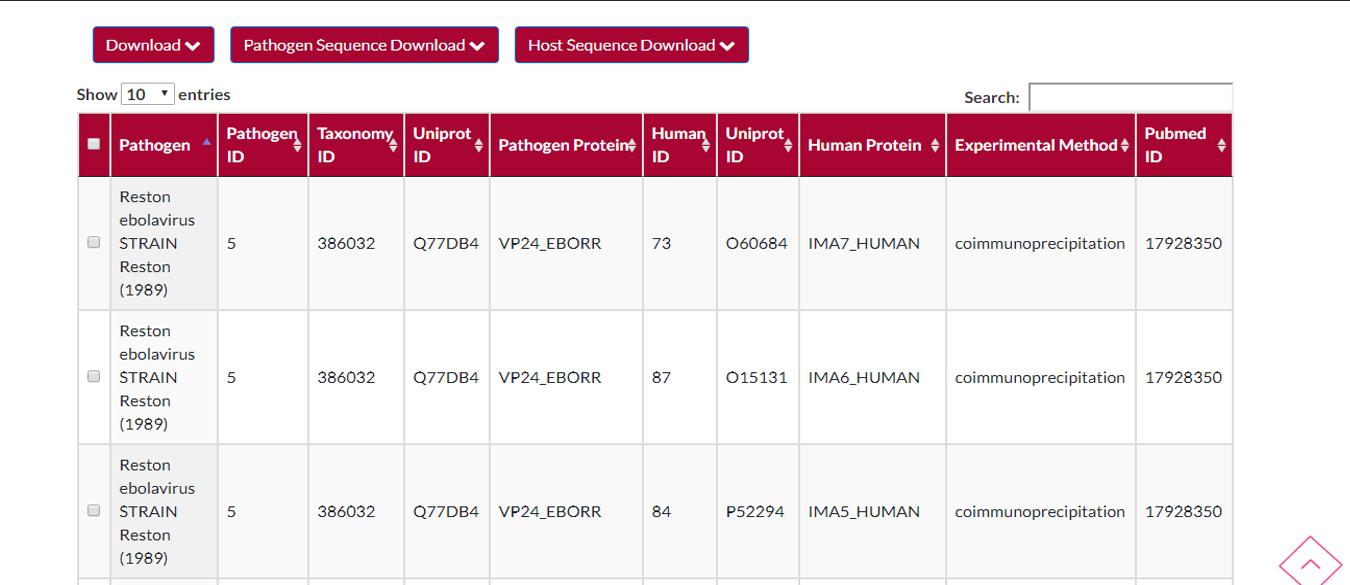
Your .xls or .xlsx file will turn into a table which has columns like (Pathogen,Pathogen ID,Taxonomy ID,Uniprot ID...).The most important thing in this table you can see that PATHOGEN ID and HUMAN ID shows the interaction of pathogen and host.
Step 4 Interaction File Upload(Table Form)
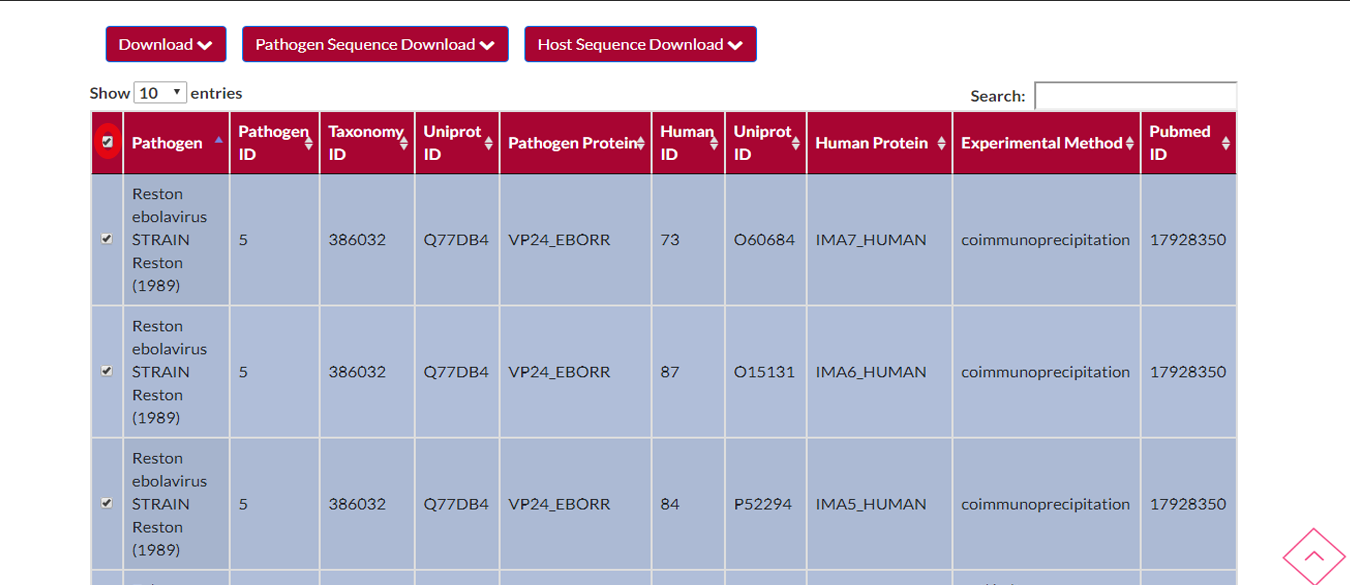
If you want to select all the rows of the active page you must check the box on the top left side of the table.
Step 1 Download
If you want to download the rows which you selected then you have to press DOWNLOAD button and select your output format that you wish for.
Step 2 Download
You may want to download the selected pathogens' protein sequences.All you have to do is to press PATHOGEN SEQUENCE DOWNLOAD button and after that a menu will appear.You can either choose to download selected pathogens or download all pathogens in the file.
Step 3 Download
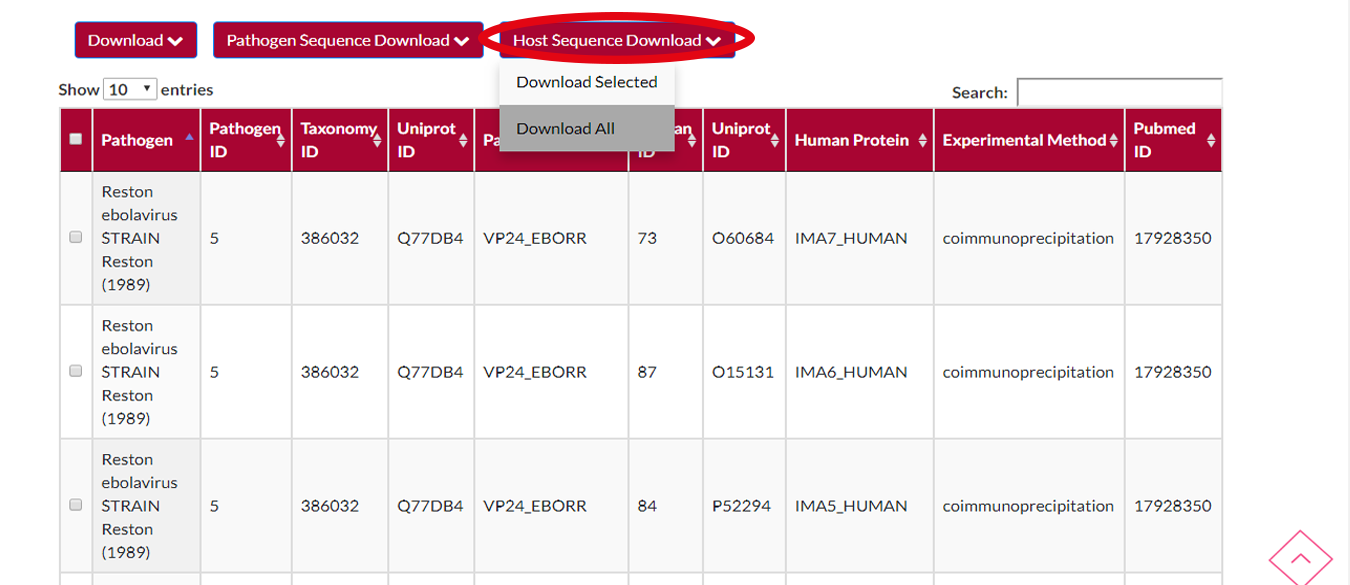
You may want to download the selected hosts' protein sequences.All you have to do is to press HOST SEQUENCE DOWNLOAD button and after that a menu will appear.You can either choose to download selected hosts or download all hosts in the file.
Step 1 Uploading the file you want to convert
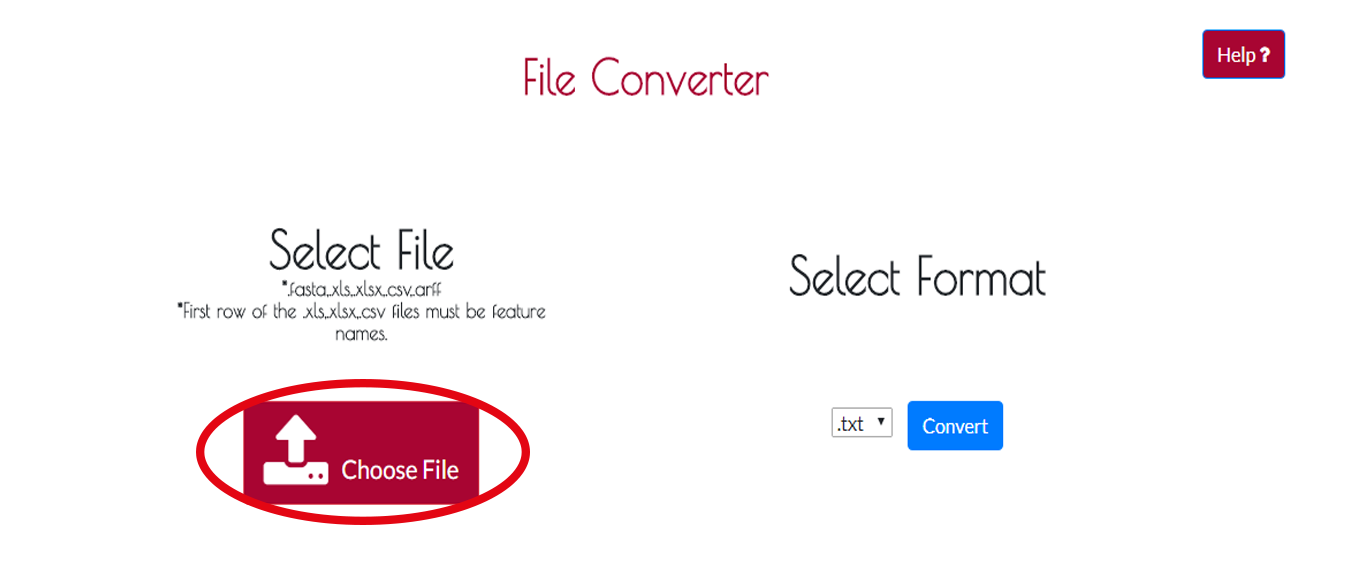
You can convert your .fasta,.xls,.xlsx,.csv,.arff files to .txt,.xls,.csv,.arff formats.First of all you have to upload your file by clicking CHOOSE FILE button.
Step 2 Uploading the file you want to convert

A navigation window will appear on the screen.Then you can find your file and press OPEN button to finish uploading process.
Step 3 The format you want to convert to
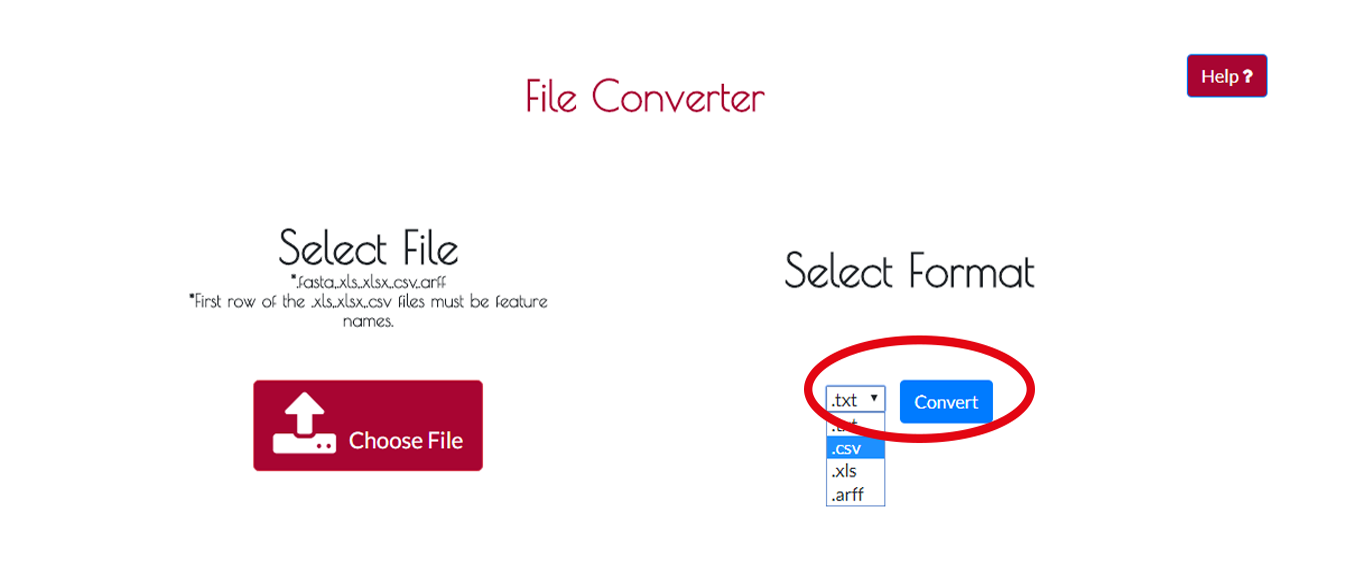
After uploading process you have to select the new format of your file.You can choose it from the drop-down menu and press CONVERT to start converting process.
Step 4 Download File
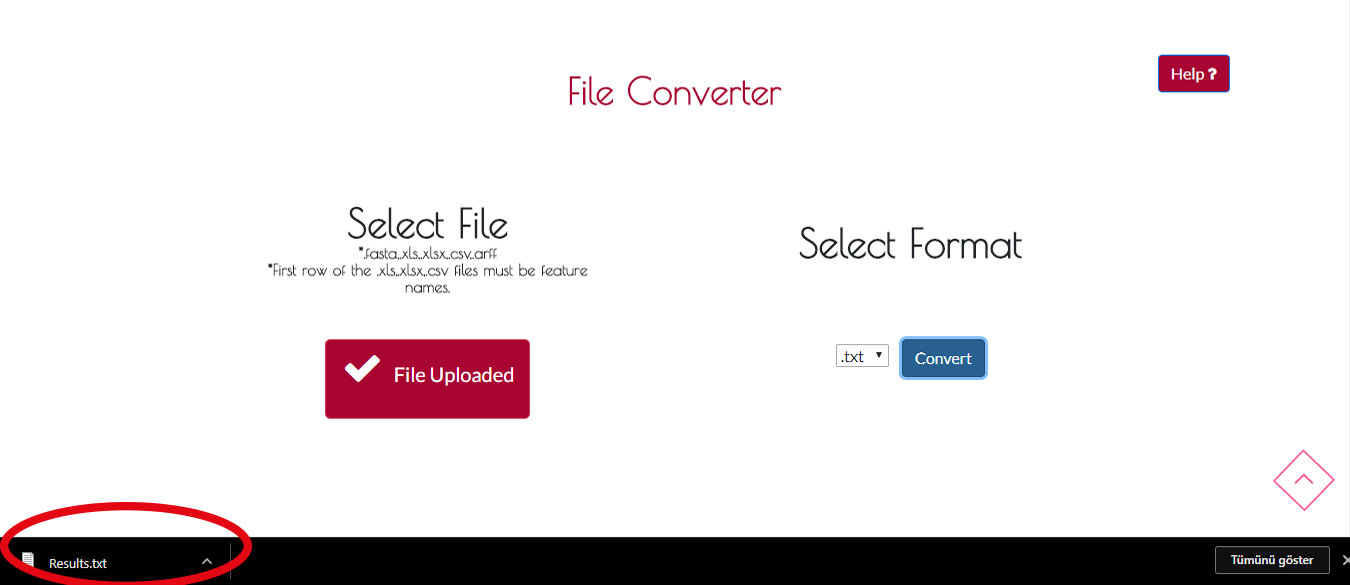
When converting process is finished , your new file will be downloaded automatically.
Step 1 Choosing the Type of Search

Select the search type by pressing the header. It will be open immediately.
Step 1 Search
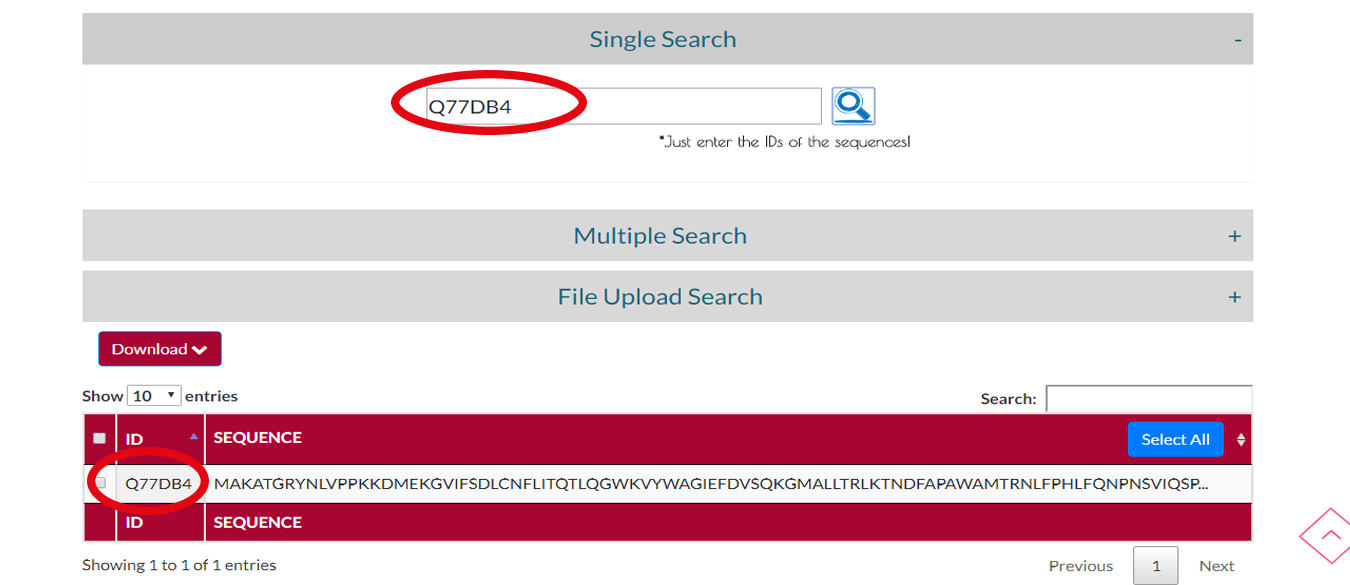
If you have an ID of a protein sequence you can choose SINGLE SEARCH to see the whole sequence of the protein.
Step 2 Search
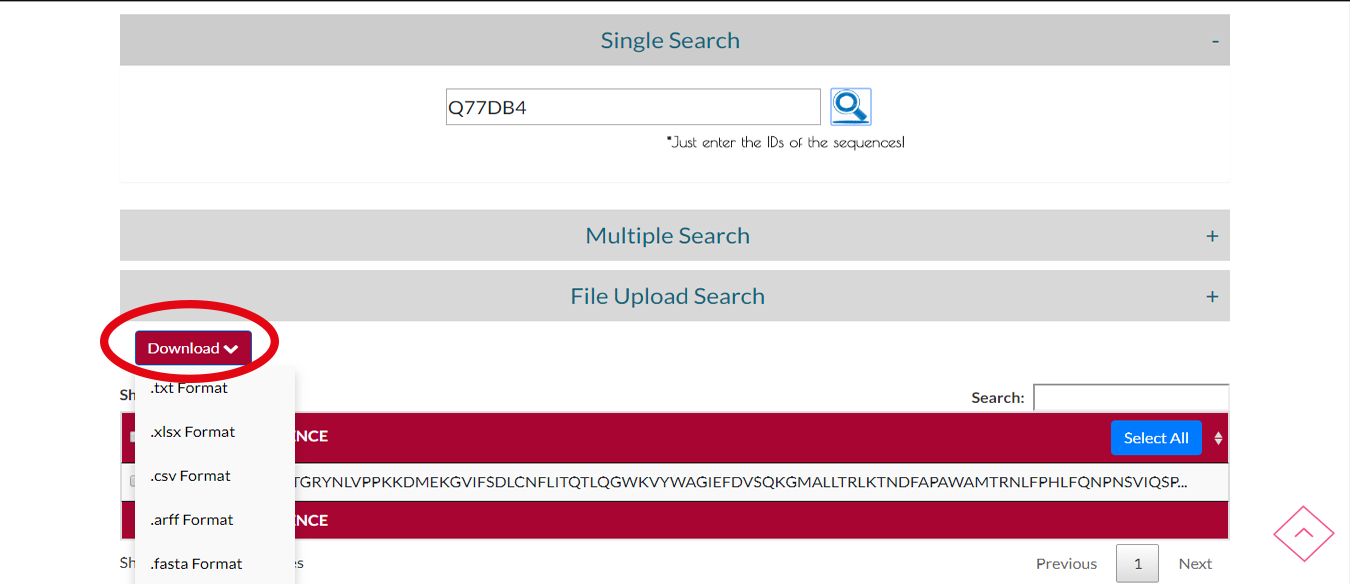
You can download the protein sequence you searched by clicking DOWNLOAD button and select the output format.
Step 1 Search
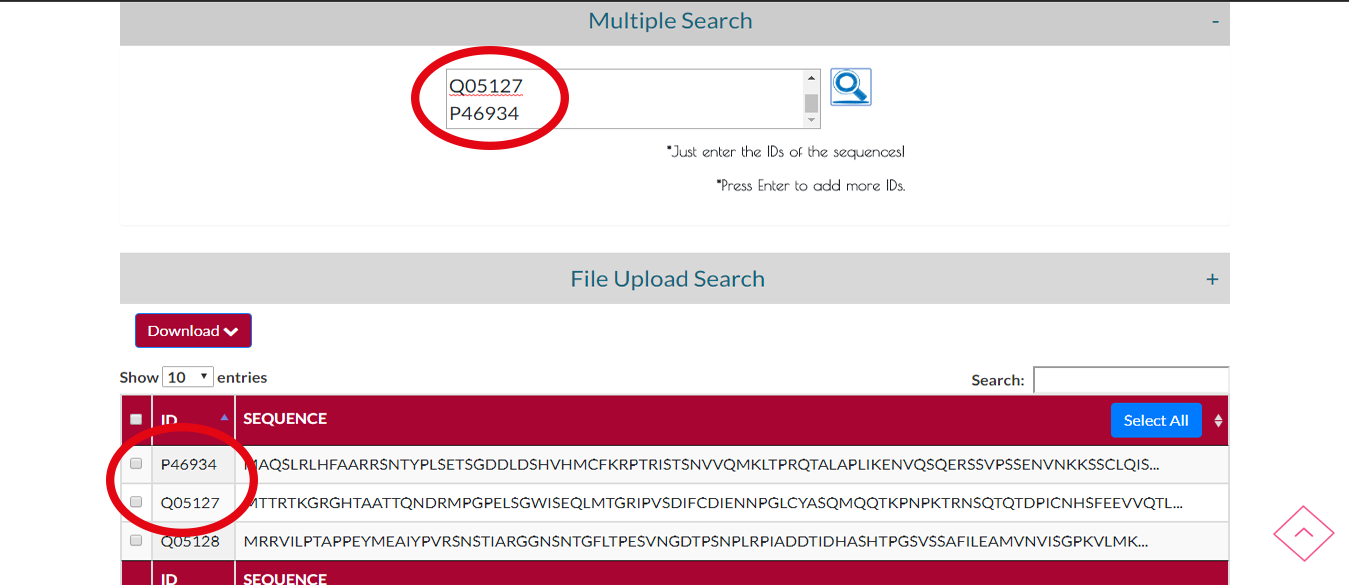
If you have lots of IDs of sequences you can choose MULTIPLE SEARCH.While you are typing IDs press enter to seperate IDs.
Step 2 Search
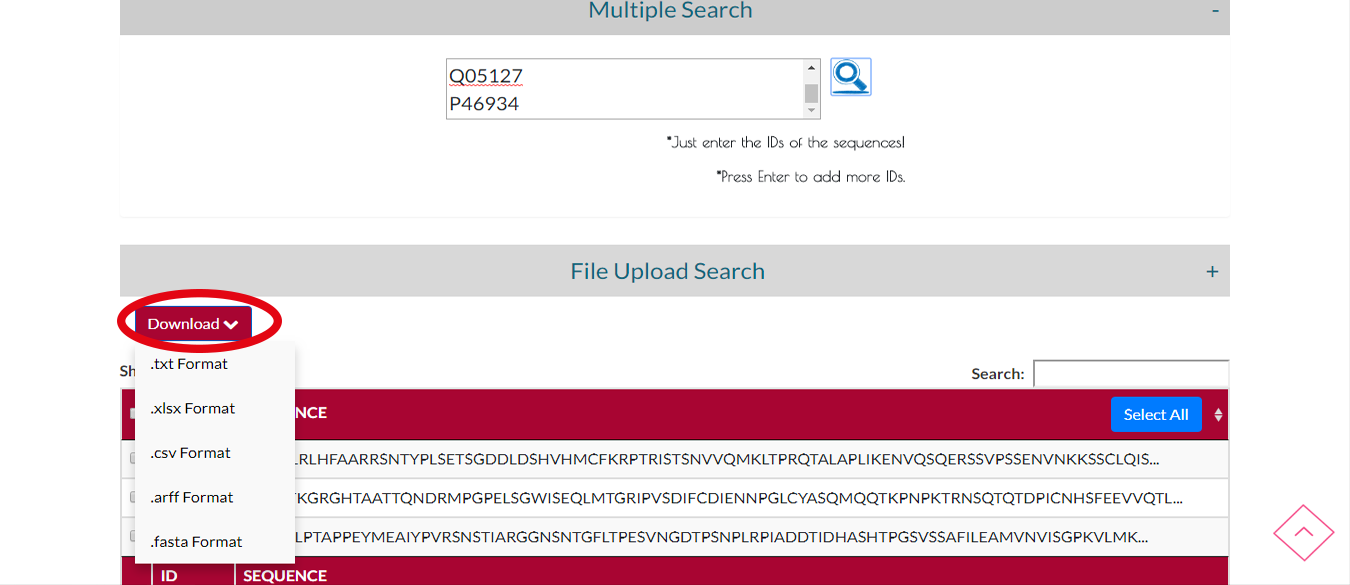
You can download the protein sequences you searched by clicking DOWNLOAD button and select the output format.
Step 1 Search
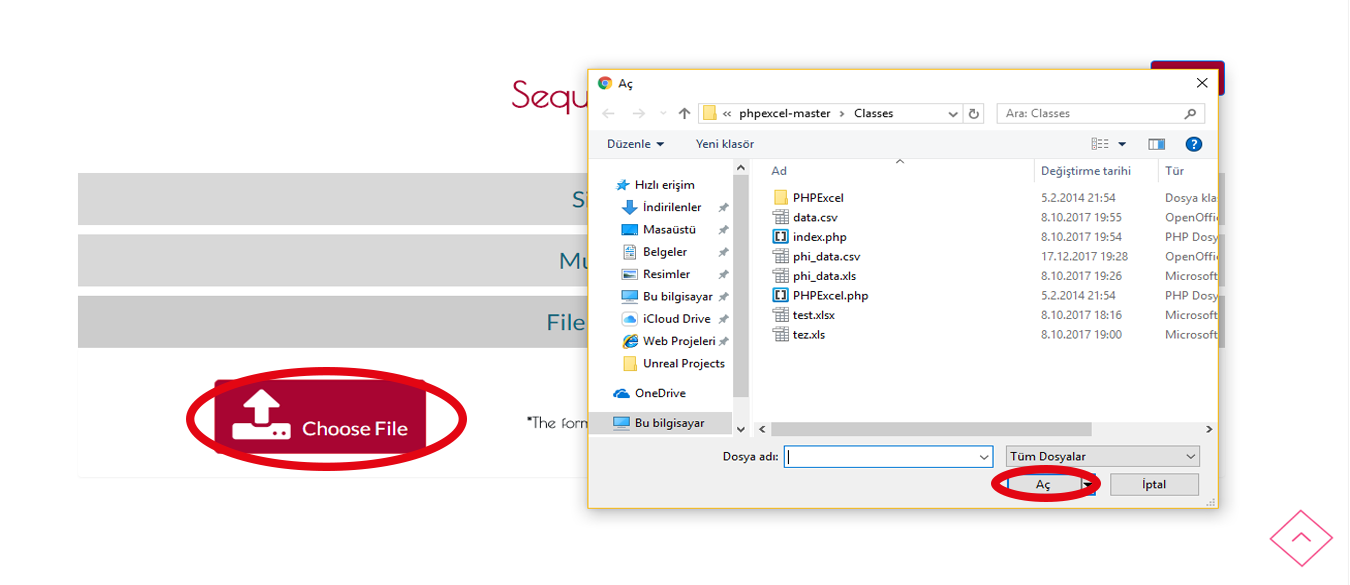
If you have .txt, .xls, .xlsx, .csv, .arff file which has only one column you can choose FILE UPLOAD SEARCH.
Step 2 Search
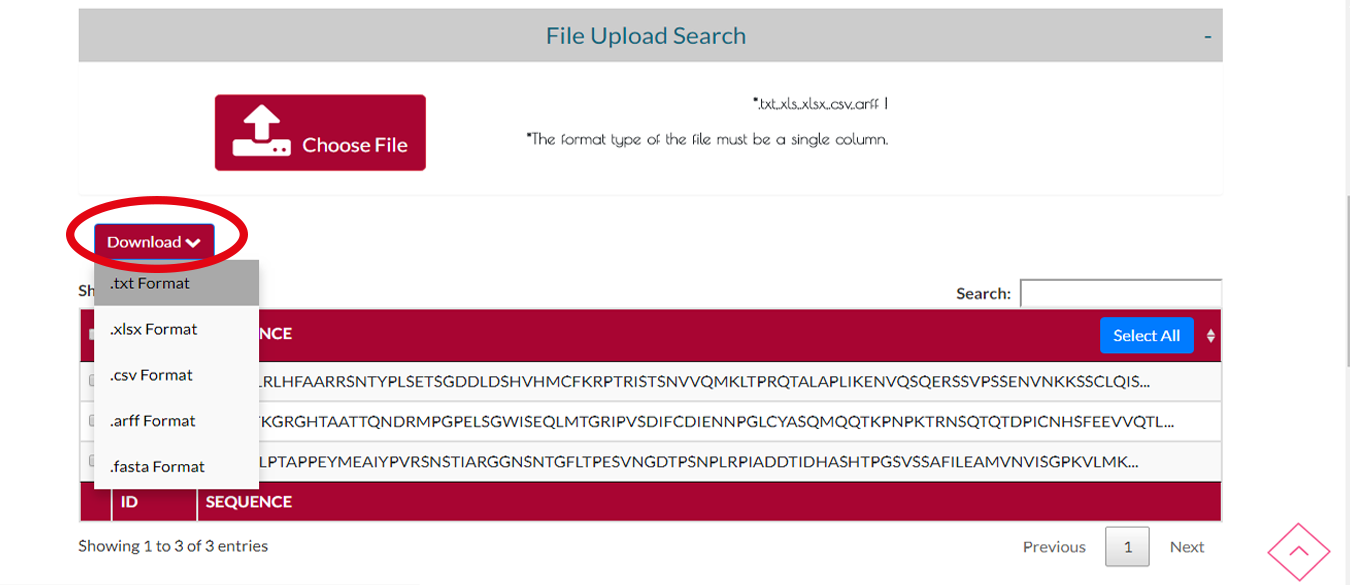
You can download the protein sequences you searched by clicking DOWNLOAD button and select the output format.

Referance : M. Bhasin and G. P. S. Raghava, “Classification of nuclear receptors based on amino acid composition and dipeptide composition,” J. Biol. Chem., vol. 279, no. 22, pp. 23262–23266, 2004.

Referance : M. Bhasin and G. P. S. Raghava, “Classification of nuclear receptors based on amino acid composition and dipeptide composition,” J. Biol. Chem., vol. 279, no. 22, pp. 23262–23266, 2004.

Referance : I. Dubchak, I. Muchnik, S. R. Holbrook, and S. H. Kim, “Prediction of protein folding class using global description of amino acid sequence.,” Proc. Natl. Acad. Sci. U. S. A., vol. 92, no. 19, pp. 8700–4, 1995.

Referance : J. Shen, J. Zhang, X. Luo, and W. Zhu, “Predicting protein–protein interactions based only on sequences information,” Proc. …, vol. 104, no. 11, pp. 4337–4341, 2007.

Referance : J. Chen, H. Liu, J. Yang, and K. C. Chou, “Prediction of linear B-cell epitopes using amino acid pair antigenicity scale,” Amino Acids, vol. 33, no. 3, pp. 423–428, 2007.

Referance : S. MAETSCHKE, M. TOWSEY, and M. BODÉN, “Blomap: an Encoding of Amino Acids Which Improves Signal Peptide Cleavage Site Prediction,” in Proceedings of the 3rd Asia-Pacific Bioinformatics Conference, 2005, pp. 141–150.

Referance : J. Guo, Y. Lin, Z. Sun, and A, "Novel Method for Protein Subcellular Localization: Combining Residue-Couple Model and SVM", in: Proceedings of Third Asia-Pacific Bioinformatics Conference, 17-21 January 2005, Singapore, 2000, vol. pp, pp. 117–129.
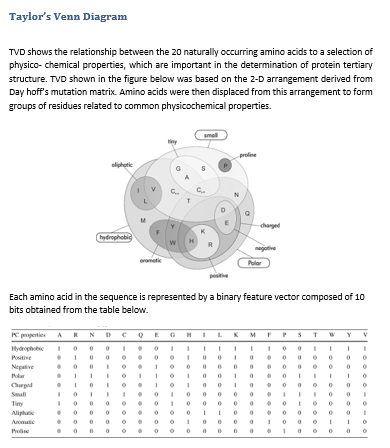
Referance : W. R. Taylor, “The classification of amino acid conservation,” J. Theor. Biol., vol. 119, no. 2, pp. 205–218, Mar. 1986.

Referance : M. Gök and A. T. Özcerit, “A new feature encoding scheme for HIV-1 protease cleavage site prediction,” Neural Comput. Appl., vol. 22, no. 7–8, pp. 1757–1761, 2013.

Referance : J. Ruan, K. Wang, J. Yang, L. A. Kurgan, and K. Cios, “Highly accurate and consistent method for prediction of helix and strand content from primary protein sequences,” Artif. Intell. Med., vol. 35, no. 1–2, pp. 19–35, 2005