Yardım
Adım 1 Dosya Yükleme

ENCODING linkine tıkladıktan sonra bu sayfaya yönlendirileceksiniz.Encoding işlemi için iki seçeneğiniz var.Protein zincirlerinizi manuel olarak girebilir veya bir .fasta dosyası yükleyebilirsiniz.
Adım 2 Dosya Yükleme
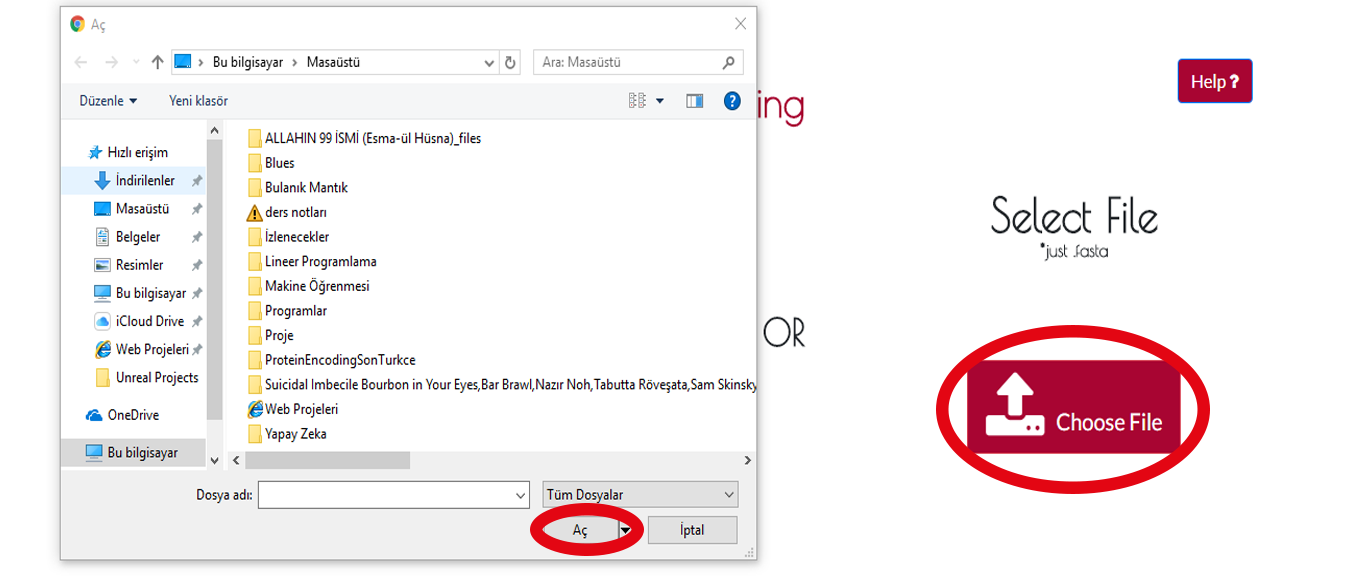
Eğer bir .fasta dosyası kullanmak istiyorsanız DOSYA YÜKLE butonuna tıklayarak dosyanızı yükleyebilirsiniz.Bu adımdan sonra bir pencere açılacak ve .fasta dosyanızı seçmeniz istenecektir.Sonra AÇ butonuna tıklayarak bir sonraki aşamaya geçebilirsiniz.
Adım 1 Zincir Girdisi

Eğer protein zincirini elle girmek isterseniz bunu textarea'ya yazmanız gerekmektedir.Eğer birden fazla zincir girmek isterseniz zincirler arasında enter a tıklamalısınız.Siz zincirleri yazarken encoding metodları belirecek ve HESAPLA metodu bir anda ortaya çıkacaktır.
Adım 2 Zincir Girdisi
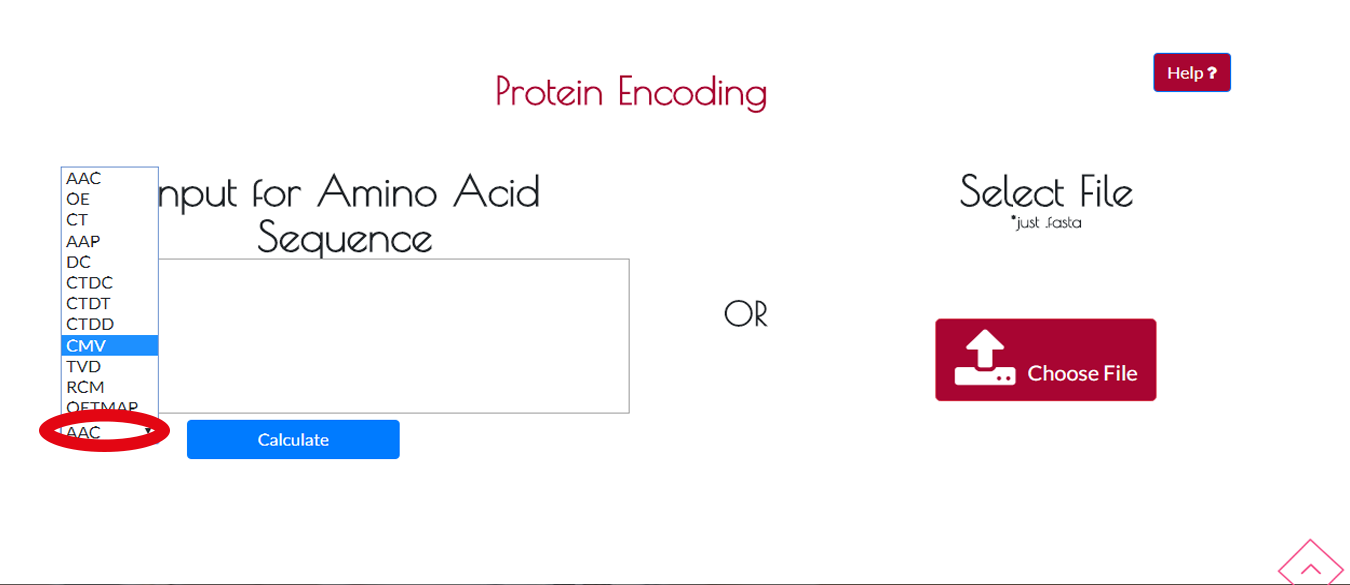
Gelen menüden encoding metodunu seçebilir ve HESAPLA butonuna tıklayarak encoding işlemini başlatabilirsiniz.
Adım 3 Zincir Girdisi
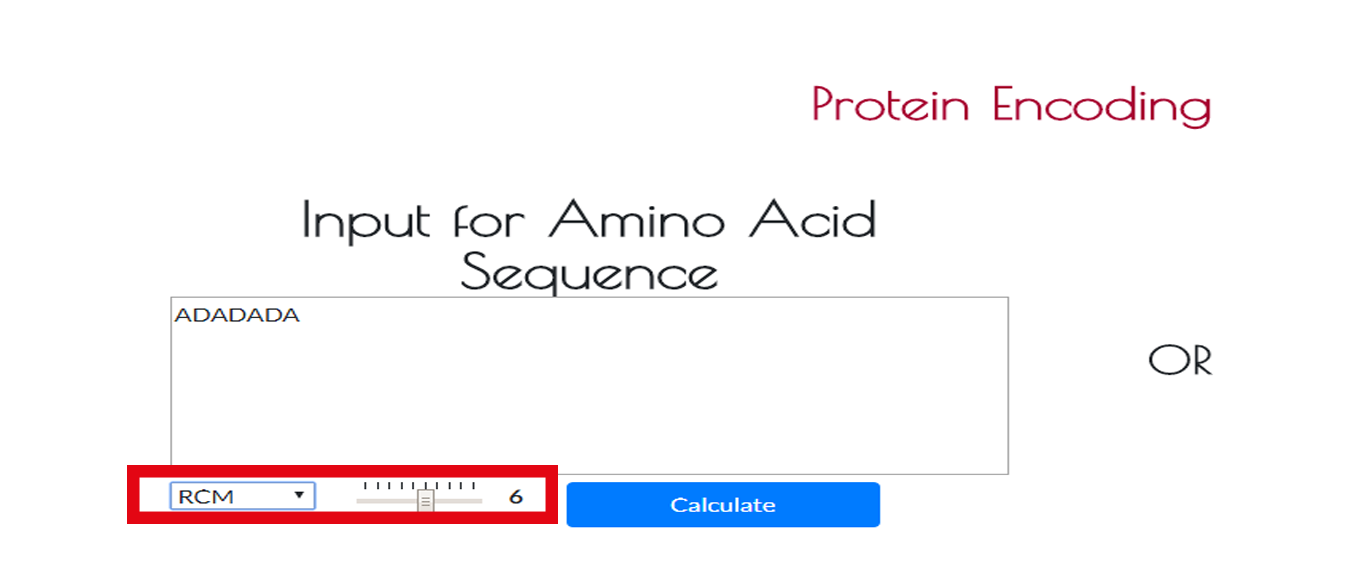
Eğer RCM metodunu seçerseniz,önce RCM metosunun rankını belirlemelisiniz.
Adım 1 Tablodan Zincir Seçimi
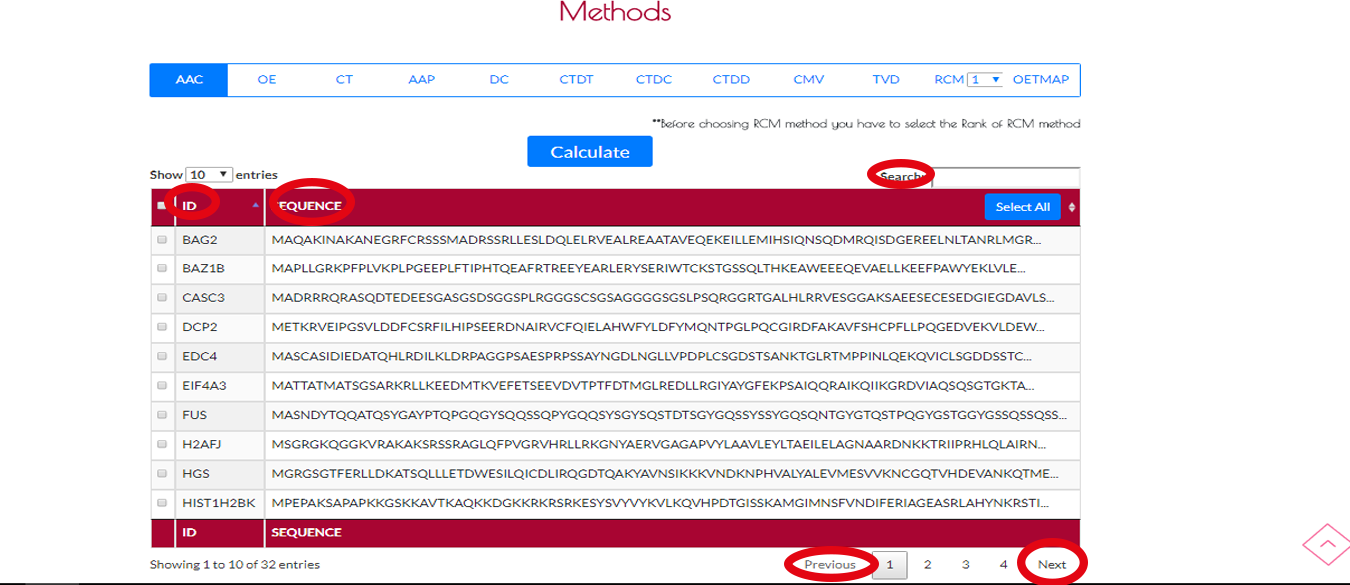
Yükleme işlemini gerçekleştirdikten sonra ID ve protein zincirleriyle dolu bir tablo ekranda beliricektir.Tablonun sağ tarafında arama seçeneğimiz mevcuttur.Bununla aradığınız ID'yi veya protein zincirini bulabilirsiniz.Tablonun alt tarafında ÖNCEKİ VE SONRAKİ butonları mevcuttur.Bu butonları kullanarak tabloda gezinebilir ve .fasta dosyanızdaki bütün ID'lere ve protein zincirlerine erişebilirsiniz.
Adım 2 Tablodan Zincir Seçimi
ablonun sağ tarafında protein zincirlerine ait checkboxlar mevcuttur.İşleme sokmak istediğiniz zincirleri manuel olarak burdan seçebilirsiniz.
Adım 3 Tablodan Zincir Seçimi
Eğer .fasta dosyasındaki bütün protein zincirlerine işlem yaptırmak isterseniz HEPSİNİ SEÇ butonuna tıklamanız gerekmektedir.Eğer bulunduğunuz sayfadaki bütün zincirleri seçmek isterseniz o zaman tablonun sol üst koşesindeki kutucuğu check etmeniz gerekmektedir.
Adım 4 Tablodan Zincir Seçimi
SelectAll butonuna tıkladığınızda buton UnselectAll olarak değişecektir.
Adım 1 Hesaplama

Protein zincirlerini seçtikten sonra , encoding metodunu seçebilir ve HESAPLA butonuna tıklayarak encoding işlemini başlatabilirsiniz.
Adım 2 Hesaplama

Eğer RCM metodunu seçerseniz,önce RCM metosunun rankını belirlemelisiniz.
Adım 3 Hesaplama

Encoding işleminden sonra sonuç tablosunu görüceksiniz.
Adım 4 Hesaplama

Son olarak sonuçları indirmek isterseniz, İNDİR butonuna tıklayabilirsiniz.Bir menü belirecek ve bu menüden çıktı formatınızı seçebileceksiniz.
Adım 1 Etkileşim Dosyası Yükleme

İlk olarak iki tip formata sahip olabilen .xls veya .xlsx dosyalarınızı yüklemelisiniz. İlk format phisto.org sitesinden indirebildiğiniz patojen-host etkileşim dosyası ve ikinci format ise sırasıyla;pathogen uniprotID,host uniprotID isimli sütünlar içeren dosyalardır.
Adım 2 Etkileşim Dosyası Yükleme
Bir pencere belirecek ve bu pencereden dosyanızın yolunu belirttikten sonra AÇ butonuna tıklayarak yükleme işlemini sonlandıracaksınız.
Adım 3 Etkileşim Dosyasını Yükleme(Tablo Formu)

.xls veya .xlsx uzantılı dosyanız sütünları (Pathogen,Pathogen ID,Taxonomy ID,Uniprot ID...) olan bir tabloya dönüştürülecek.Bu tablodaki en önemli ayrıntı görüceğiniz üzere PATOJEN ID ve KONAK ID lerinin patojen-konak etkileşimlerini gösterdiğidir.
Adım 4 Etkileşim Dosyasını Yükleme(Tablo Formu)

Eğer bulunduğunuz sayfadaki bütün satırları seçmek isterseniz tablonun sol üst köşesindeki kutucuğu check etmeniz gerekmektedir.
Adım 1 İndirme

Eğer seçtiğiniz satırları indirmek isterseniz o zaman İNDİR butonuna tıklamalı ve istediğiniz çıktı formatını seçmelisiniz.
Adım 2 İndirme

Seçtiğiniz patojenlerin protein zincirlerini indirmek isteyebilirsiniz.Tek yapmanız gereken PATOJEN ZİNCİRİ İNDİR butonuna tıklamak ve ilgili menünün açılmasını beklemek.Seçili patojenleri indirmeyi seçebilirsiniz veya bütün dosyadaki patojenleri indirmeyi de seçebilirsiniz.
Adım 3 İndirme

Seçtiğiniz konakların protein zincirlerini indirmek isteyebilirsiniz.Tek yapmanız gereken KONAK ZİNCİRİ İNDİR butonuna tıklamak ve ilgili menünün açılmasını beklemek.Seçili konakları indirmeyi seçebilirsiniz veya bütün dosyadaki konakları indirmeyi de seçebilirsiniz.
Adım 1 Çevirmek İstediğiniz Dosyayı Yükleme

.fasta,.xls,.xlsx,.csv,.arff,.txt uzantılı dosyalarınızı .xls,.xlsx,.csv,.arff formatlarından herhangi birine çevirebilirsiniz.Öncelikle yüklemek istediğiniz dosyayı DOSYA YÜKLE butonuna tıklayarak siteye yüklemelisiniz.
Adım 2 Çevirmek İstediğiniz Dosyayı Yükleme

Ekranınıza bir pencere gelecektir.Bu pencereden dosyanızı bulduktan sonra AÇ butonuna tıklayarak yükleme işlemini sonlandırabilirsiniz.
Adım 3 Çevirmek İstediğiniz Format

Yükleme işlemini tamamladıktan sonra dosyanızı çevirmek istediğiniz formatı seçmelisiniz.Yeni formatı menüden seçip ÇEVİR butonuna tıkladığınızda çevirme işlemini başlatmış olursunuz.
Adım 4 Dosyayı İndirme

Çevirme işlemi bittikten sonra, yeni dosyanız otomatik olarak indirilecektir.
Step 1 Choosing the Type of Search

Select the search type by pressing the header. It will be open immediately.
Adım 1 Arama
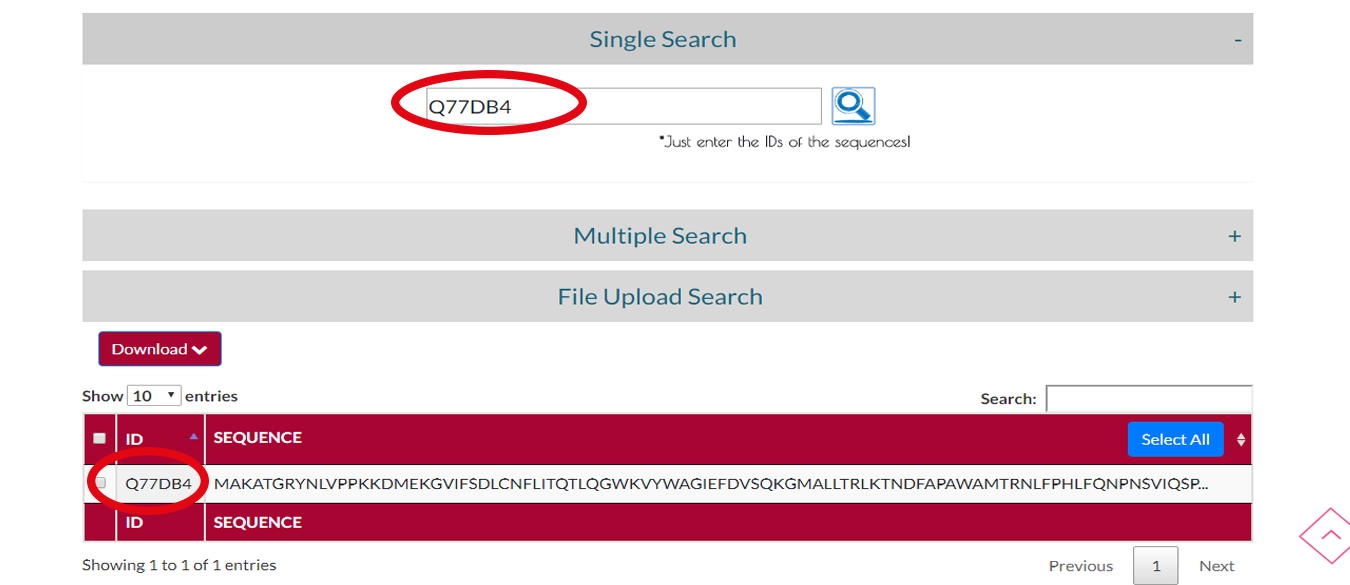
Eğer bir protein zincirine ait bir ID varsa TEKLİ ZİNCİR ARAMASINI kullanabilirisiniz.
Adım 2 Arama
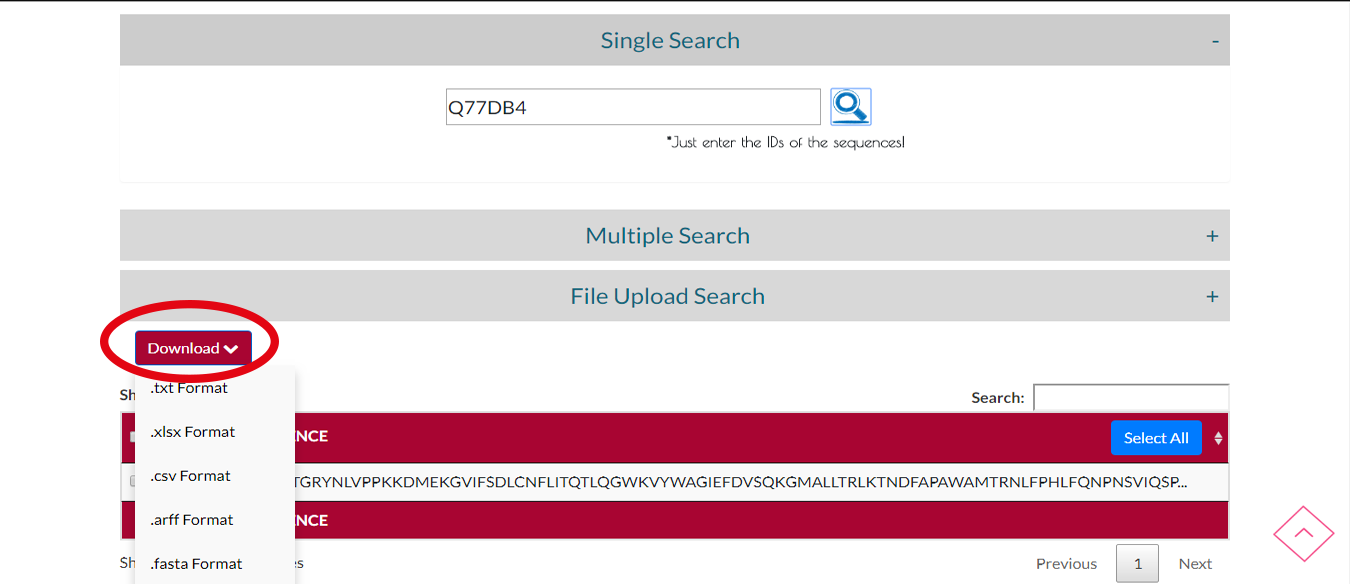
Aradığınız protein zincirini çıktı formatını seçip İNDİR butonuna tıklayarak indirebilirsiniz.
Adım 1 Arama
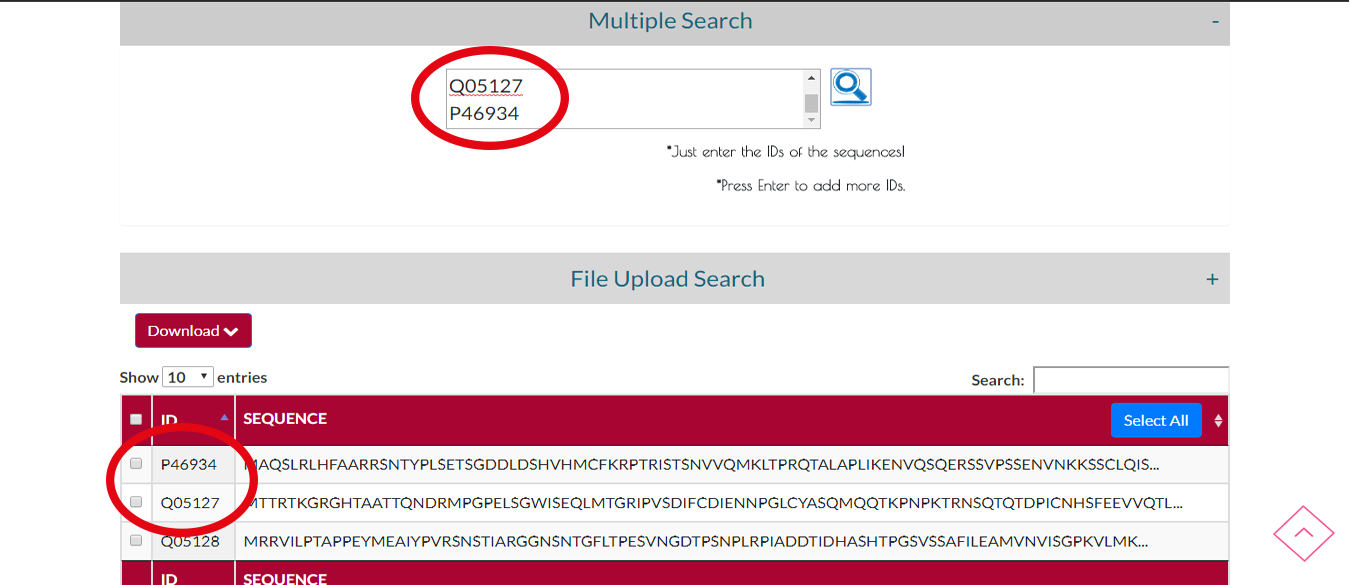
If you have lots of IDs of sequences you can choose MULTIPLE SEARCH.While you are typing IDs press enter to seperate IDs.
Adım 2 Arama
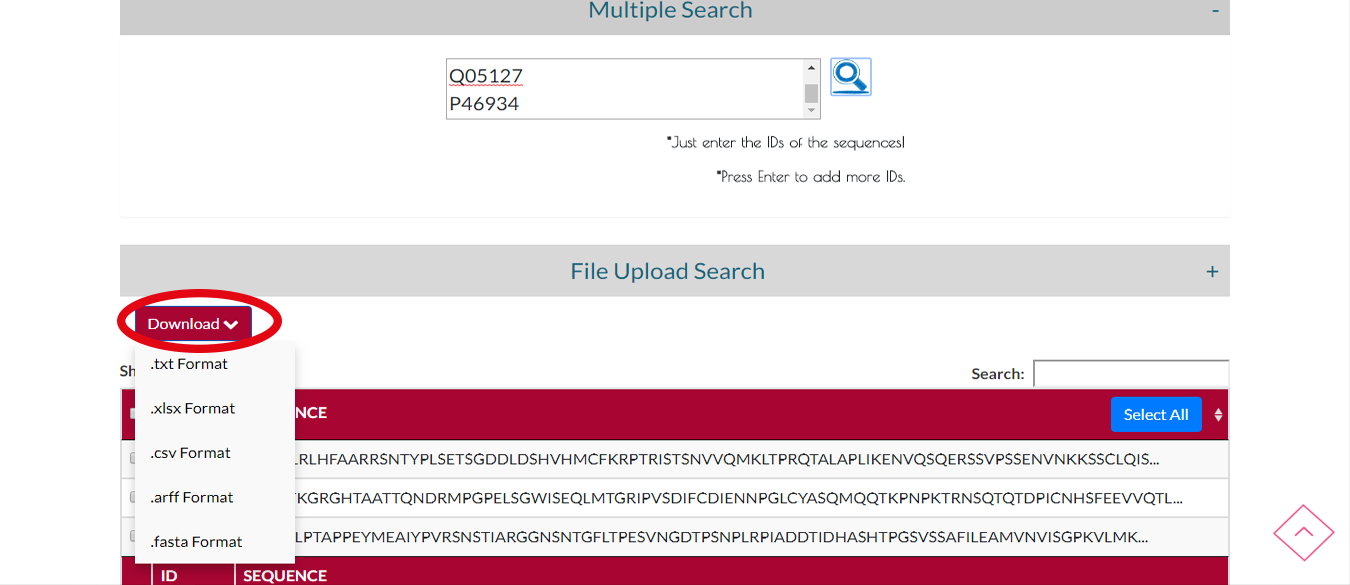
Aradığınız protein zincirlerinin çıktı formatını seçip İNDİR butonuna tıklayarak indirebilirsiniz.
Adım 1 Arama
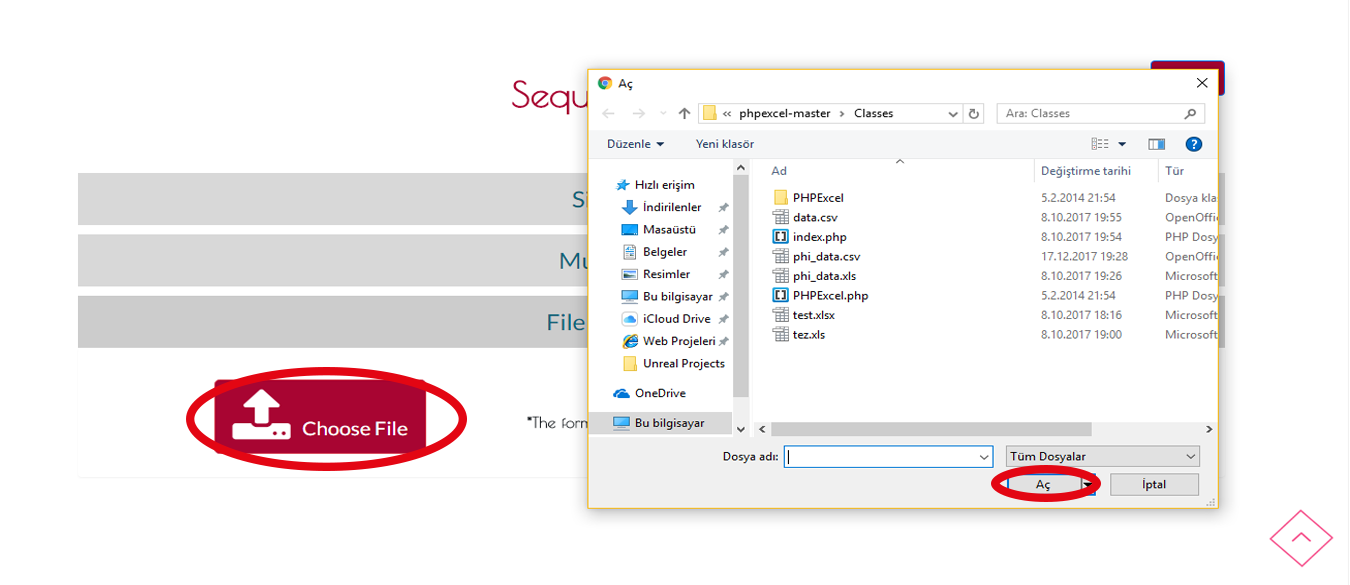
Eğer sadece tek sütüna sahip .txt, .xls, .xlsx, .csv, .arff formatlı bir dosyaya sahipseniz DOSYA YÜKLEMELİ ARAMAYI kullanabilirsiniz.
Adım 2 Arama
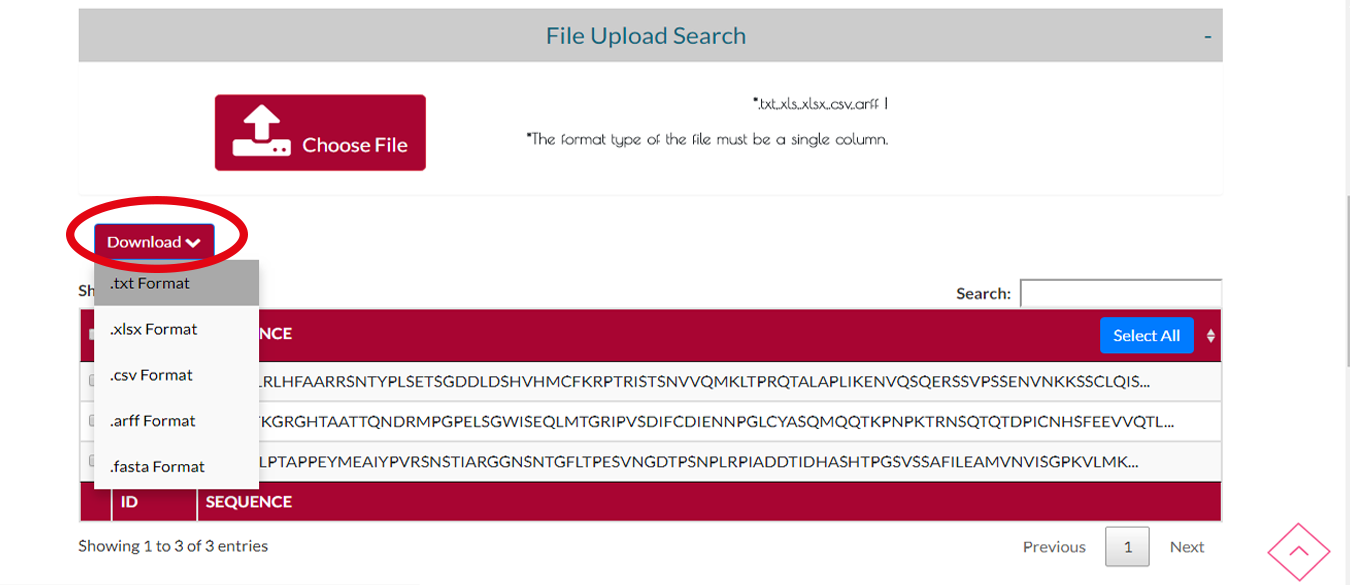
Aradığınız protein zincirlerinin çıktı formatını seçip İNDİR butonuna tıklayarak indirebilirsiniz.
Amino Acid Composition (AAC)
Amino Asit Kompozisyonu bir protein içindeki herbir amino asit tipinin oranını verir.Bir protein zinciri aşağıdaki gibi tanımlanır:
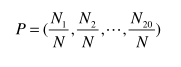
Ni i tipli amino asitin sayısını belirtirken N ise zincirin uzunluğunu belirtir.
Dipeptide Composition (DC)
Dipeptid Kompozisyonu bir zincirdeki herbir amino asit ikilisinin oranını verir.Aşağıdaki gibi tanımlanır:
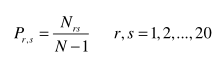
Nrs r ve s tipli amino asitleri içeren dipeptidlerin toplamını belirtirken N zincirin uzunluğunu belirtir.
Composition, Transition, and Distribution (CTDC, CTDT, CTDD)
Amino asitlerin dizilimi, kalıntıların bazı yapısal veya fizikokimyasal özelliklerinin (öznitelikleri) dizilerine dönüştürülür. Yirmi amino asit, Tomii ve Kanehisa'nın amino asit indekslerinin ana kümelerini temsil eden yedi farklı amino asit özelliklerinin her biri için üç gruba ayrılmıştır.
Bir protein içindeki üç grubun her birinin global yüzde bileşimini tanımlamak için belirli bir öznitelik için üç tanımlayıcı, kompozisyon (CTDC), geçiş (CTDT) ve dağılım (CTDD) hesaplanır; yüzde frekansları, özellik değişirken sırasıyla proteinin tüm uzunluğu boyunca dizin ve sırayla öznitelik dağılım örüntüsü.
Conjoint Triad (CTriad)
20 amino asit, dipolleri ve yan zincir hacimleri açısından 7 sınıflara ayrılır. Birleşik üçlü, her üç sürekli amino asidi bir birim olarak görür ve aynı sınıftaki kalıntılar özdeş elementler olarak tanımlanır.7 grup olduğundan dolayı 343 Conjoint üçlüsü var.İ inci birleşik üçlü şu şekilde tanımlanır:
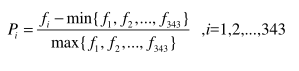
Amino Acid Pair (AAP)
Amino Asit Çifti, Chen ve arkadaşları tarafından geliştirilmiş olup diğer proteinlere karşı belirtilen fonksiyonlara sahip proteinlerde dipeptidlerin oluşumunu tanımlamaktadır. Örneğin, AEACCGCA peptidi 7 AAP'ye ayrılabilir: AE, EA, AC, CC, CG, GC ve CA. There are 20*20=400 AAPs.Bir AAP bileşeni şu şekilde tanımlanır:
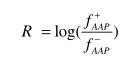
Burada + f (AAP) ve -f (AAP) belirtilen işlevlere sahip proteinlerdeki ve diğer proteinlerin belirli bir AAP'nin oluşma sıklığıdır.Daha sonra AAP ölçeği merkezileştirilir ve normalize edilir:
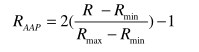
Burada Rmin, Rmax, R'nin min ve max değerleridir. Buna ek olarak, sıralı dipeptidlerin oluşumu, RAAP matrisi ile çarpım sonucu ortaya çıkar ve sonuç olarak 400-boyutlu bir vektör oluşur.
Orthonormal Encoding
Ortonormal kodlama, amino asitlerin yaygın olarak kullanılan bir temsilidir. Her amino asit türü (genel olarak 20 ortak tür), bir bitin 1 olduğu ve diğerlerinin 0 olduğu 20 bitlik bir ikili dize ile temsil edilebilir.Böylece, bir N amino asit dizisi, 20 * N boyutlarında bir vektöre dönüştürülür.
Residue-Couple Model (RCM)
Geleneksel hücre altı yer tahmin modeli esas olarak amino asit kompozisyon modeline dayanır. Bununla birlikte, amino asit kompozisyon modeli tek başına protein dizisinin belirli bir miktarını yoksayar. Ne yazık ki, dizi sırası hakkındaki bilgi, olası ardışık düzen sayısından ötürü tahmin için bir model tanıma işine kolayca dahil edilemez (Chou, 2001). Bununla birlikte, Chou'nun sözde sıralama modeli ve Yuan'ın Markov zinciri modelinden esinlenerek, dolaylı olarak dizi düzen efektini kullanan yeni bir model geliştirdik.
Model, bir dizi protein serisini harf dizisi olarak gösterir:
R1R2 R3 R4 R5 R6 R7 …… RL
burada Ri, l (I= 1,2, ..., L) konumundaki amino asidi temsil eder. "Artık çift" aşağıdaki gibi tanımlanmıştır:
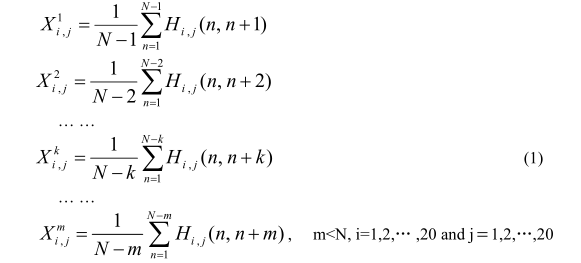
burada, n konumundaki amino asit i iken ve konum n + k'deki amino asit ise j ise, Hi, j (n, n + k) = 1; Aksi takdirde, Hi, j (n, n + k) = 0 (Şekil 1). I ve j değerleri 20 farklı amino asiti temsil eden 1 ila 20 arasında değişir (kısaca A, C, D, E, F, G, H, I, J, K, L, M, N, P, Q, R, S, T, V, W, Y). Xi1, j (i, j = 1,2, ..., 20), bir protein sırasındaki sürekli kalıntı çiftlerinin bir modunun gözlemlendiği sıklığı temsil eden çift 1. dereceli kalıntı-çift olarak adlandırılır. ("_" herhangi bir amino asit türünü temsil eder) bağlanma modunun (i, _, j) gözlemlendiği frekansı temsil eden 2. dereceli kalıntı-çift olarak adlandırılır; Birleşmiş modun (i, _, _, j) bir protein dizisinde gözlemlendiği frekansı temsil eden çiftler ve benzeri. Bu nedenle, her sırada 20 × 20 = 400 kalıntı-çift vardır (Şekil 1).
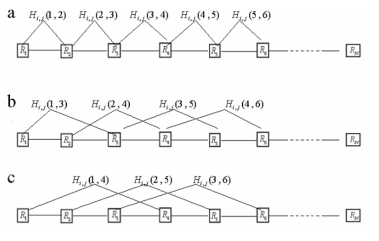
Şekil 1: Farklı rütbe ile artık-çifti gösteren şematik bir çizim: (a) birinci dereceden: birbirini izleyen iki ardışık arasındaki bağlanma modu. (b) 2. sıra: aralarında sadece bir amino asit olan iki kalıntı arasındaki bağlanma modu. (c) 3. sırada: aralarında sağ iki amino asit bulunan iki kalıntı arasındaki bağlanma modu.
Her bir protein sırası için, tüm kalıntı çiftler bir x vektörüne birleştirildi, yani x'in ilk 400 bileşeni 400 birinci derece kalıntı çift oldu ve aşağıdaki bileşenler 400 ikinci seviye kalıntı çifti ve vb. Bu nedenle, son vektörün boyutu 400 × m'dir. M'nin değerine, "birleştirme derecesi" denir ve çiftlerin toplam sırasını temsil eder. Bu model hem amino asit bileşimi hem de protein dizisinin düzen etkisi için bilgi içerir. Her protein dizisi, 400 x m boyut vektörleri kümesi elde etmek için bu şekilde analiz edilir (her vektör bir vektöre tekabül etmektedir). Vektör seti, eğitim ve tahmini için destek vektör makinesine girdi vektörleri olarak kullanılmıştır(Şekil 2).
Taylor's Venn Diagram (TVD)
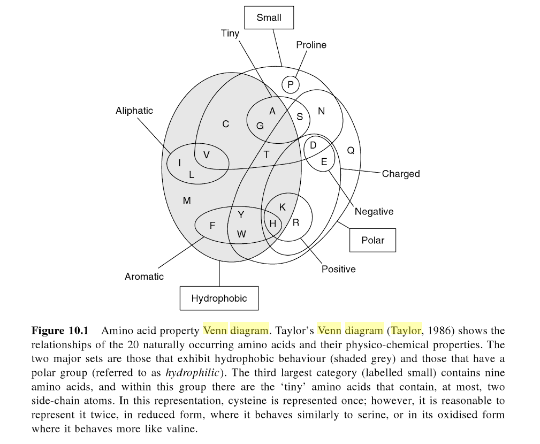
Amino asit özellik Venn diyagramı,Taylor Venn Diyagramı 20 amino asitin ilişkilerini ve fizikokimyasal özelliklerini göstermektedir.İki büyük kümeden biri hydrofobik diğeride polar (hydrofilik) dır.Üçüncü büyük grup 9 amino asit ve içlerinde küçük amino asitler olan bir grupda vardır.
OETMAP
OE, dizilerin gösterimi için ortak bir yöntemdir. Amino asit dizilerinden ikili olarak elde edilen tüm vektörlerin karşılıklı ortogonal ve boyut bakımından eşit olmasını sağlar. Fakat OE, proteinler hakkında bilgi ve dizi homolojisine sahip değildir. Birlikte lokalize olan amino asit dizilerinin, amino asit fizikokimyasal özelliklerine göre bazı benzerlikler paylaşması gerektiğine inanıyoruz. TVD, doğal olarak bulunan 20 amino asit arasındaki ilişkiyi, protein üçüncül yapısının belirlenmesinde önemli olan fiziko-kimyasal özelliklere göre gösterir. Şekil 1'de gösterilen TVD, Day hoff'un mutasyon matrisinden türetilmiş 2-D düzenlemeye dayanıyordu. Amino asitler daha sonra, bu fizikokimyasal özelliklerle ilgili artık grupları oluşturmak için bu düzenlemeden çıkarıldı. TVD'nin HIV-1 özgüllük problemi için amino asitler arasındaki fizikokimyasal ilişkiden ayrı olduğuna inanıyoruz. Yani, amino asitlerin benzer fizikokimyasal tipleri, bölünebilir bir peptid için bir yerde oluşur. Fakat TVD, amino asit gösterimi için yeterli değildir. Sonuç olarak geliştirdiğimiz OETMAP, birbirlerini tamamlayan OE ve TVD yöntemlerinin birleşiminden oluşmaktadır.
P, HIV-1 proteaz klivaj veri kümesindeki bir amino asit sekansı olsun. Pi, P'deki i'inci amino asittir (i = 1, 2, ..., L için L, amino asit dizisinin uzunluğudur).
Her bir Pi'ye karşılık gelen, {ai} ve {b i} özellik vektörlerini aşağıdaki gibi oluşturduk:
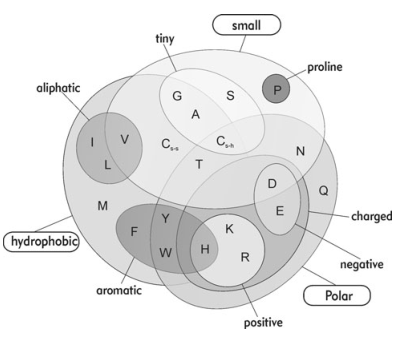
{ai}, Pi için OE vektörü (20 bit) 'dür. I'nin değeri, 20 farklı amino asiti (kısaca A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V).
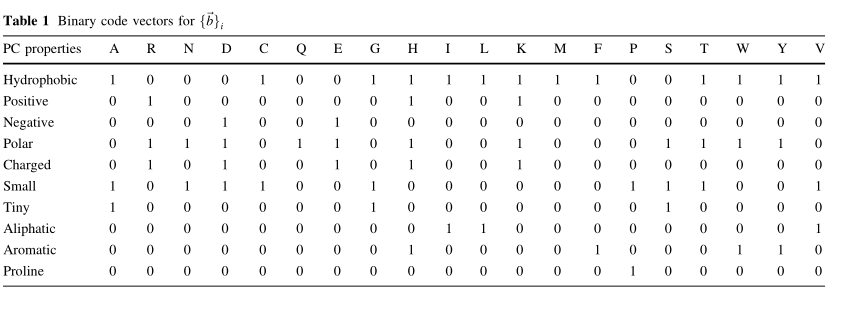
bi, Tablo 1'de gösterilen, TVD'den elde edilen 10 bitten oluşan ikili bir özellik vektörüdür.
Yapılan özellik vektörleri {ai} ve {bi} P i araya getirilir:
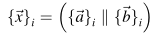
Son olarak, 30 L boyutuna sahip P'nin özellik vektörü X, aşağıdaki gibi sırayla gösterilir:
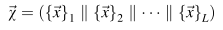
Yeni tedbiri açıklamak için HIV-1 veri setinde ALDFEQEM dizisi için aşağıdaki hesaplama örneği gösterilmektedir. Peptid dizisindeki D kalıntısı için OE eşlemesini aşağıdaki gibi hesapladık:

Bu hesaplama prosedürleri, özellik vektörü X'ü elde etmek için peptid dizisinin her bir amino asidi için tekrarlanmaktadır. OETMAP'in tüm dizinin üzerinde kullanılması yüksek boyutluluk gerektirdiğinden bunu azaltmak için, bir asal bileşim analizi (PCA) uyguladık.Verinin varyans miktarının maksimum dağılımı ve lineer diskriminant analizi (LDA) ile optimal alt uzayı arayan ve sınıflar arası dağılımı en yükseğe çıkarabilen ve sınıf içi dağılımı simültane bir şekilde en aza indirebilen, verinin düşük boyutlu gösterimi. PCA ve LDA her ikisi de en popüler çizgisel boyut azaltma teknikleridir.
Rognvaldsson ve siz, HIV-1 proteaz bölünmesinin doğrusal bir problem olduğunu ve iki sınıf desen sınıfını ayrıştırmak için maksimum marj hiper düzlemini bulmayı amaçlayan LSVM'nin, bu problem için en iyi bilinen sınıflandırıcı olduğunu gösterdiniz. Bu nedenle, LSVM, yaptığımız testlerde HIV-1 proteaz bölgelerini öngörmek için benimsenmiştir.
Composition Moment Vector (CMV)
Bir protein sınıflandırma görevi için in vitro tahmini yöntemlerin zaman alıcı olması ve masraflı olması nedeniyle, model tanıma / makine öğrenme algoritmalarına dayanan hesaplama yöntemleri kullanılır.Tam bir model tanıma sistemi, sınıflandırılacak gözlemleri toplayan bir sensör, ön işleme modülü gürültüyü ortadan kaldırır, deseni normalleştirir, desendeki sayısal bilgileri kodlayan / seçen bir özellik kodlama / seçim mekanizması ve sınıflandırma görevini uygulayan bir sınıflandırma algoritması.
Özellik kodlama süreci, orijinal gösterim alanından sınıfların daha kolay ayrılabilir yeni bir alana eşlemesini sağlar. Özellik kodlamanın amacı, model verilerini sınıflayıcı algoritmalarına mümkün olduğunca tanımlamaktır. CMV özellik kodlaması yöntemi amino asit terkibi ve bir dizi sırasındaki pozisyon bilgisini hesaba katar. Diğer bir deyişle CMV, dizideki amino asidin bileşimi ve konumu ile yapı içeriğiyle fonksiyonel ilişki hakkında bilgi içerir, yani farklı yapı içeriğine ama aynı moment vektörüne sahip iki veya daha fazla primer amino asit sekansı olmamalıdır.Üstelik, birincil dizideki her AA hakkında bilgi sağladığı için diğer önlemlerden daha kapsamlı bir açıklaması verir.